
Human Papillomavirus (HPV) Viral Proteins Substitute for the Impact of Somatic Mutations by Affecting Cancer-Related Genes: Meta-analysis and Perspectives
Zisheng Shang1, Valentina L. Kouznetsova2,3, Igor F Tsigelny2-5*
1MAP Program UC San Diego, La Jolla, California, USA
2Moores Cancer Center UC San Diego, La Jolla, California, USA
3San Diego Supercomputer Center, UC San Diego, La Jolla, California, USA
4Department of Neurosciences, UC San Diego, La Jolla, California, USA
5CureMatch Inc. San Diego, California, USA
Abstract
Purpose: Although a strong association between human papillomavirus (HPV) and a variety of cancers has long been established, infection by HPV alone has been shown to be insufficient for the induction of cancer, with a large number of HPV infections regressing without causing cancer. Additionally, HPV-negative cases have worse prognosis rates than HPV-positive ones across a multitude of cancer types. The reasons behind these phenomena are poorly understood. We try to explain it.
Methods: In this meta-analysis study, we first compared the mutation sets of HPV-positive and HPV-negative cancers using data from the Cancer Genome Atlas (TCGA) and from open published sources. Then, focusing on the genes that are exclusively mutated in HPV-negative head and neck cancer, we used KEGG pathways and reviewed literature to investigate how HPV can mimic the effects of mutations in those genes and thereby trigger carcinogenesis despite the absence of those mutations.
Results: Using TCGA mutation frequencies of significantly mutated genes stratified by HPV status, we extracted 17 genes exclusively mutated in HPV-negative cancers and detailed the HPV infection mechanisms that are largely able to mimic the effects of mutations in 13 of these genes. Through our literature searches and pathway analyses, we also presented antitumor effects caused by HPV infection that reduce carcinogenic efficiency and could contribute to the prognosis difference.
Conclusion: Our findings confirm that HPV infection substitutes for some, but not all, somatic driver mutations required for the induction of cancer by mimicking the effects of those mutations through various virus–host interaction mechanisms, thereby accelerating the cancer-related pathways.
Introduction
The human papillomavirus (HPV) is a DNA virus that infects the skin and mucus membranes and has long been identified as a common etiological agent for many types of cancers in those types of tissues1. In fact, it is associated with almost 100% of cervical cancers, 90% of anal cancers, a significant percentage of head-and-neck squamous cell carcinomas, 40% of vulva, vagina, and penile cancers, 12% of pharyngeal cancers, and 3% of oral cancers1. It has also recently been shown to be associated with bladder carcinomas2,3. Among all the subsites of head-and-neck cancer (which include the larynx, oral cavity, hypopharynx, oropharynx and the sinonasal tract), cancers in the oropharynx presently show the most pronounced link to HPV infection4,5.
At least 200 types of HPV have been identified, but only a few are truly oncogenic and are classified as high-risk types of HPV, which include the well-known HPV-16 and HPV-18 because of their high association with squamous cell carcinomas6. Additionally, among the multiple viral proteins that HPV encodes, E6 and E7 stand out as the main viral proteins that support carcinogenesis, as they drive cellular proliferation, block apoptosis and inhibit the cell cycle arrest mechanisms7. These mechanisms either accelerate the rate of cell division or inhibit the normal regulations on the cell-cycle-progression system, thus leading to unchecked cell growth, which may lead to cancer.
However, despite HPV’s high prevalence in squamous cell carcinomas, study results have shown that HPV infection alone is not sufficient for the induction of cancer. In fact, the majority of HPV infections are asymptomatic and regress: about 70% of new HPV infections resolve spontaneously in one year and 90% naturally resolve in two years8. Progression to cancer is a rare event: only in a small percentage of cases do these viral infections persist and cause epithelial lesions, which increase the risks of cancer development but are still not definite indicators of cancer9,10,11. So, why are these outcomes of HPV infection drastically different from each other? What factors besides HPV infection contribute to these differences?
Another interesting phenomenon exists in the difference in prognosis between HPV-positive and HPV-negative cancers. For oropharyngeal cancers, compared to HPV-negative patients, HPV-positive patients tend to be younger, with less exposure to tobacco and alcohol, and there is a higher incidence of HPV-positivity for males than females12. Across a multitude of tumor sites, HPV-positive cancers are associated with younger patient ages and are shown to confer better survival rates4,13. Many previous studies have correlated the better survival rates with younger ages and less tobacco and alcohol exposure, dismissing the role of HPV as an important prognostic factor. However, there are studies that contradict this assumption, finding that the prognosis differences still exist after adjusting for the age, smoking and drinking prognostic factors14,15.
Overall, to explain the substantial difference between possible HPV infection outcomes and the observation that HPV-positive cancers often occur earlier in life than negative ones, we hypothesize that HPV infection mimics the effects of some somatic driver mutations by targeting a set of genes that play key roles in various carcinogenic pathways. Note that this set of genes might differ from the normally mutated driver genes in HPV-negative cases. This hypothesis implies that HPV infection has to be accompanied by the appropriate somatic driver mutations in order to support cancer development, while the mere infection itself is insufficient, because HPV doesn’t mimic the effects of all the mutations necessary to support cancer development. Such mimicking mechanism reduces the number of driver mutations required for the induction of cancer for HPV+ patients, because HPV infection can substitute the long somatic mutation accumulation process. We also hypothesize that this mimicking effect is not as powerful as the carcinogenic effects of a necessary set of random somatic driver mutations, possibly explaining the observation that HPV-positive status often confers better prognosis in HPV-associated cancers.
We explored the possibility of our hypothesis mainly by investigating the differences in necessary mutation sets between HPV-positive and HPV-negative cases and by analyzing the effects of HPV on genes that are exclusively mutated in HPV-negative cases.
Materials and Methods
KEGG Pathway Database
Short for the Kyoto Encyclopedia of Genes & Genomes, the KEGG database16 contains pathway diagrams that graphically illustrate many biochemical pathways. The pathways are all constructed based on existing reliable literature. In this study, we mainly used the KEGG pathways titled “Pathways in Cancer—Homo Sapiens” and “Human Papillomavirus Infection” to guide our investigation of mutation effects and HPV viral effects.
The Cancer Genome Atlas (TCGA)17
TCGA, started in 2006, is a landmark cancer genomics program that catalogued the genetic mutations responsible for a variety of cancer types. In this study, we used head-and-neck squamous cell carcinoma mutation frequency data that is stratified by HPV status from The Cancer Genome Atlas. This data serves as the basis for our investigations, as it provides the exclusively mutated genes in HPV-negative cases.
Results
HPV-negative cases require more somatic driver mutations than HPV-positive cases
In cancers associated with HPV, HPV-negative cases generally occur with the presence of significantly more somatic driver mutations than HPV-positive ones. Using gene mutation frequency data stratified by HPV status from The Cancer Genome Atlas17, we have constructed a table to compare the mutation frequencies of significantly mutated genes in HPV-positive and HPV-negative cases of head-and-neck cancer (Figure 1; data used is summarized in Table 1). The genes in the dataset play key roles in the cell death, immunity, differentiation, and oxidative stress signaling pathways including Ras/Raf and Akt/PI3K. To extract the “validated” somatic mutations, a threshold line is placed at 3% mutation frequency, as suggested by Ma et al.18. After taking this threshold, we noticed that there were 17 genes exclusively mutated in HPV-negative cases (and not mutated in HPV-positive cases), while only 4 genes were exclusively mutated in HPV-positive cases (with 0% mutation frequency in HPV-negative cases). This 17/4 ratio may demonstrate that more driver mutations are needed to induce cancer in HPV-negative cases than HPV-positive cases. As we previously proposed, this difference could be due to HPV proteins activating or inhibiting the specific genes, substituting for the random somatic mutation accumulation process.
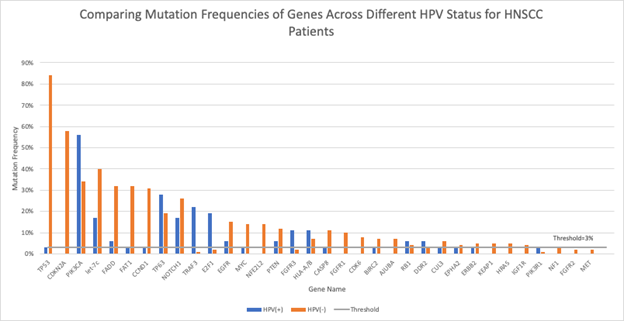
Figure 1: HNSCC Gene Mutation Frequencies by HPV Status
Comparisons of mutation frequencies of genes playing key roles in several important cancer pathways for HPV-positive (blue) and HPV-negative (orange) head-and-neck squamous cell carcinoma (HNSCC) patients. The threshold at 3% mutation frequency is indicated by the horizontal gray line.
Table 1. HNSCC Mutation Frequencies by HPV Status Data Table.
Important genes in several key cancer pathways and their mutation frequencies in HPV-positive (HPV+) and HPV-negative (HPV–) cases17. Genes that are exclusively mutated in HPV-negative cases (after taking the threshold at 3%) are in bold.
|
Gene |
HPV+ Mutation Frequency |
HPV– Mutation Frequency |
|
TP53 |
3% |
84% |
|
CDKN2A |
0% |
58% |
|
PIK3CA |
56% |
34% |
|
let-7c |
17% |
40% |
|
FADD |
6% |
32% |
|
FAT1 |
3% |
32% |
|
CCND1 |
3% |
31% |
|
TP63 |
28% |
19% |
|
NOTCH1 |
17% |
26% |
|
TRAF3 |
22% |
1% |
|
E2F1 |
19% |
2% |
|
EGFR |
6% |
15% |
|
MYC |
3% |
14% |
|
NFE2L2 |
0% |
14% |
|
PTEN |
6% |
12% |
|
FGFR3 |
11% |
2% |
|
HLA-A/B |
11% |
7% |
|
CASP8 |
3% |
11% |
|
FGFR1 |
0% |
10% |
|
CDK6 |
0% |
8% |
|
BIRC2 |
3% |
7% |
|
AJUBA |
0% |
7% |
|
RB1 |
6% |
4% |
|
DDR2 |
6% |
3% |
|
CUL3 |
3% |
6% |
|
EPHA2 |
3% |
4% |
|
ERBB2 |
3% |
5% |
|
KEAP1 |
0% |
5% |
|
HRAS |
0% |
5% |
|
IGF1R |
0% |
4% |
|
PIK3R1 |
3% |
1% |
|
NF1 |
0% |
3% |
|
FGFR2 |
0% |
2% |
|
MET |
0% |
2% |
In addition to the scenario in head-and-neck cancer mentioned above, we noticed a similar situation occurring in cervical cancer. According to a study conducted by Banister and co-authors19 that analyzes cervical cancer data from The Cancer Genome Atlas, “HPV-active tumors had on average 115 somatic mutations per tumor, whereas HPV-inactive tumors had 228 somatic mutations per tumor.” Furthermore, by comparing the somatic mutation frequencies of 26 key cervical cancer driver genes between HPV-positive and HPV-negative cases using two-tailed Fisher’s Exact p-values and obtaining the odds ratios, Banister and colleagues19 showed that all 26 genes had different mutation rates across the different types of cases, and that all of the genes were more frequently mutated in HPV-negative cases (Figure 2), as the odds ratios of somatic mutations in HPV-negative vs. HPV-positive tumors are greater than 1 for all listed genes. These findings further support the notion that HPV-negative cancers require more somatic driver mutations than HPV-positive ones, across different HPV-associated cancer types.
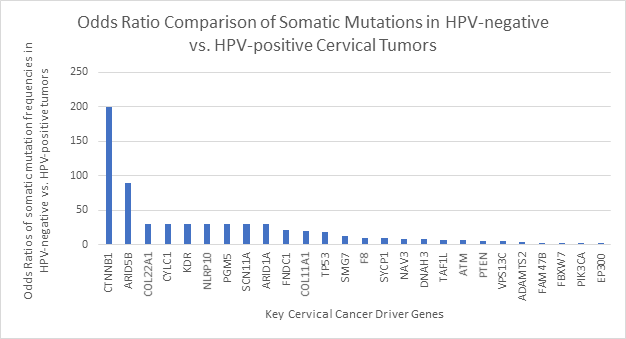
Figure 2: Odds Ratio Comparison of Somatic Mutations in Cervical Tumors by HPV Status.
Somatic mutation differences between HPV-inactive (HPV-negative) and HPV-active (HPV-positive) cervical cancers, as shown through odds ratios of somatic mutation frequencies in HPV-negative vs. HPV-positive cancers for 26 key cervical cancer driver genes19. All odds ratios are larger than one, indicating that for all of the genes listed the mutation frequencies are higher in HPV-negative cases than in HPV-positive cases. The data used for the construction of the figure is extracted from the study of Banister et al.19.
We also note that in a study conducted by Iranzo and co-authors20 to estimate the average number of driver mutations needed for various types of cancers, the number of driver mutations needed for the induction of cervical cancer is only about 1, with a range from about 0.1 to 1.8. This surprisingly small number probably accounts for the extremely high HPV-prevalence in cervical cancers. Thus, corresponding to our proposed idea that the required number of driver mutations in HPV-positive patients are generally less than the number in HPV-negative patients, the fact that almost all cervical cancers are supported by HPV makes the average number of driver mutations surprisingly small.
Prognosis Stratified by Age Group & HPV Status
As mentioned above, HPV-positive status generally confers better prognosis than HPV-negative status in a wide range of HPV-associated cancers, including HNSCC, in which the presence of HPV generally leads to lower risks of dying, lower risks of recurrence and better response to therapy21,22. Previous interesting hypothesis addressing this scenario has associated such prognosis outcome with the ages of HPV-positive and HPV-negative patients23-26. As Larsson and colleagues23 corrected the overall and recurrence-free survival rates for age and tumor size, they discovered that the presence of HPV doesn’t confer a survival advantage anymore and concluded that HPV isn’t an independent prognostic factor in the diagnosis of cancer. This hypothesis seems reasonable, as many studies showed that HPV-positive cancers generally occur earlier in life than HPV-negative cancers27-29, and this younger age for HPV-positive cancers would confer better prognosis30.
However, there exist published data that calls into question the validity of this hypothesis. As shown in Figure 3(A), within each age group, the HPV-positive 5-year survival rate is better than that of HPV-negative cases for cervical cancer14. In Figure 3(B), the hazard ratios (HRs) show the risk of death for HPV-positive patients compared with HPV-negative patients of oropharyngeal cancer, which is a common type of head and neck cancer15. Both unadjusted and adjusted HRs are shown, and adjusted HRs are adjusted for sex, race, income, education, Charleson comorbidity index, facility type, insurance status, year of diagnosis, primary tumor subsite, treatment, and American Joint Committee on Cancer Eighth Edition clinical T stage, clinical N stage, and M stage. When HR=1, the two groups have the same risk of death; when HR>1, HPV-positive patients are at a greater risk of death, and when HR<1, HPV-positive patients are at a lower risk of death. From these data, we can see that both adjusted and unadjusted HR<1 for all age groups, thus HPV-positive patients are always at a lower risk of death than HPV-negative patients. Therefore, HPV status is an independent prognostic marker, as the positive effect of its presence is certain regardless of the age group. There is an exception in the <30 years old age group for cervical cancer, where the HPV-negative category demonstrates a 5-year relative survival rate of 100%. However, such a result is probably due to the extremely small number of data points available within that age group.
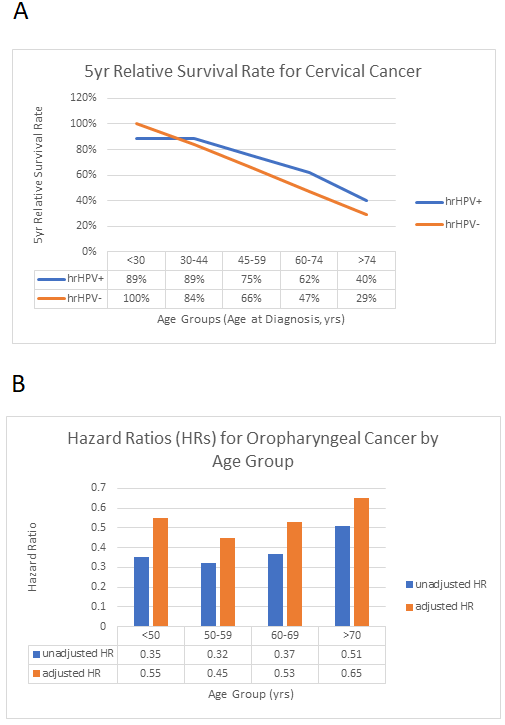
Figure 3: Prognosis Stratified by Age Group and HPV Status.
A. 5-year relative survival rate for cervical cancer, stratified by age group and HPV status. The figure is based on the data of Lei et al.14. We see that the 5-year relative survival rates for high-risk HPV-positive patients are higher than those for high-risk-HPV-negative patients except for the age group <30, which may have yielded skewed results because of the extremely small number of available data points. B. Hazard Ratios (HRs) for Oropharyngeal Cancer by Age Group (age groups are <50, 50-59, 60-69 and >70). The figure is based on the data of Rettig et al.15. From these hazard ratios, we can see that for oropharyngeal cancer, HPV-positive patients are at a lower risk of death than HPV-negative patients across all age groups.
HPV infection partially mimics the somatic driver mutation accumulation process
We propose that the significant difference between the number of somatic driver mutations required for the induction of cancer in HPV-positive and -negative cases is accounted for by the HPV viral infection mechanisms. In the absence of some necessary driver mutations, HPV proteins target a set of genes that activate or inhibit certain cancer pathways to support cancer development, mimicking the effects of those somatic driver mutations. This mimicking mechanism could only be partial and thus could account for the prognosis difference between HPV-positive and -negative cancers. In other words, the HPV infection might not be as effective in inducing cancer as the driver mutations.
To investigate and evaluate the proposal above, we focused on the genes that were exclusively mutated in HPV-negative cases after the threshold. We analyzed KEGG pathways16 and performed public sources analysis to see how HPV infection mechanisms could substitute for these mutations and cause cancer in their absence. We also analyzed the differences between the effects of mutation and infection to look for specific factors that could contribute to the prognosis difference between HPV-positive and -negative groups. According to Figure 1 and Table 1, there are 17 of the exclusively mutated genes: TP53, CDKN2A, FAT1, CCND1, MYC (c-MYC), NFE2L2, CASP8, FGFR1, CDK6, BIRC2 (cIAP), AJUBA, CUL3, EPHA2, ERBB2, KEAP1, HRAS, and IGF1R.
TP53
TP53 is the gene coding for the P53 protein, the mutation of which is present in a wide variety of tumor cells. Most TP53 mutations are missense mutations that occur in the central sequence-specific DNA-binding domain and inactivate the tumor suppressor’s transcriptional regulation functions31. Such inactivation and loss of function affect a significant number of P53 transcriptional targets including cell cycle inhibitors like P21, apoptosis related proteins such as BAX, and DNA-repair proteins like GADD45 (Figure 4A)32-34.
These effects of mutation-induced inactivation of P53 can be mimicked by HPV proteins through targeting of a set of genes ultimately linked to P53 or the P53 pathway. First of all, the E6 viral oncoprotein can bind with E6AP, and this E6/E6AP complex directly associates with P53. This association allows E6AP to catalyze the transfer of ubiquitin to P53, thereby activating the proteasome-mediated degradation of this tumor-suppressing protein. Thus, E6 inhibits P53’s ability to transcriptionally activate a number of proteins playing key roles in cell cycle regulation. For example, it inhibits the activation of P21, preventing P21 from causing G1 cell cycle arrest through its binding and inhibition of CDK2. This interaction between HPV E6 and P53 is widely studied over the last several decades and is common to HPV infections in a variety of cancer types35,36. Secondly, the E6 oncoprotein can bind to and inhibit p300/CBP, which normally activates P53-dependent transcription. Thus, this inhibition can repress the transcriptional activity of P5337. However, this binding and inhibition has been noted by Zimmermann et al.37 to be limited to E6 proteins of high-risk HPVs associated with cervical cancer. Finally, in cervical infections, HPV E6 inactivates TADA3 function, abrogating TADA3’s role as a coactivator for P53-dependent transcriptional activation and disrupting the cell cycle arrest mechanism following DNA damage38.
Independent from its abnormal transcriptional functions, mutant P53 induces a series of oncogenic gain-of-function mechanisms, which are functions independent from and unrelated to the wild type P53 protein. Studies on Li-Fraumeni syndrome patients, who are subject to the effects of mutant P53, reveal that mutant P53 can increase genomic instability through its disruption of normal spindle checkpoint control, generating polyploidy cells39. It has also been shown by studies of myeloid leukemic cells that mutant P53 can confer antiapoptotic advantages to tumor cells, rendering cells significantly more resistant to cMyc and a number of other anticancer agents40,41. In addition, in studies performed on non-small cell lung carcinoma cells, with the cooperation of oncogenic Ras, mutant P53 activates the expression of a large set of chemokines and interleukins that have been shown to induce metastasis and angiogenesis42,43. While the gain-of-function effects of mutant P53 are observed in the above-stated non-HPV-associated cancers, mutant P53 could carry out similar effects in HPV-associated cancer types, and HPV seems to be unable to mimic all these gain-of-function effects of oncogenes induced by mutant P53. Thus, the mutant P53 in HPV-negative cases could have increased oncogenic effects when compared to P53 that is altered by HPV.
On the other hand, the E7 HPV protein, although commonly known for its carcinogenic effects when interacting with the retinoblastoma protein, produces antitumor effects with its interactions with wild type P53. In a study using mouse embryo fibroblasts, Seavey and colleagues44 showed that the E7 viral protein increased the level of P53 protein by inhibiting the interaction between P53 and MDM2, so that MDM2 cannot degrade P53 anymore. Such an inhibition minimizes the effects of E7/PP2A and E5/V-ATPase interactions, which were supposed to degrade P53 through the activation of MDM2. Specifically, without its inhibitory function, E7 would cause the degradation of P53 by directly binding to PP2A to displace its B subunit. This binding would leave PP2A unable to dephosphorylate and inhibit Akt, which is one of PP2A’s targets. As a result, high levels of Akt would enhance the activation of MDM2, which monoubiquitinates P53 and thus mediates P53’s degradation by proteasomes45,46. The overactivation of MDM2 would then result in degradation of P53 and thus cell-cycle dysregulation. However, this carcinogenic effect is inhibited by E7’s inhibition of MDM2–P53 interactions. Also, without this inhibitory function of E7, HPV E5 would use MDM2 to trigger P53 degradation. In transfected human keratinocytes, E5 has been shown to interact with the subunit C (a structural component) of V-ATPase to impair the enzyme’s functions47. Such an impairment triggers a series of events in the PI3K-Akt signaling pathway (Figure 4B) and ultimately leads to overactivation of MDM248. However, the effect is seriously mitigated by E7’s inhibition of MDM2-P53 interaction.
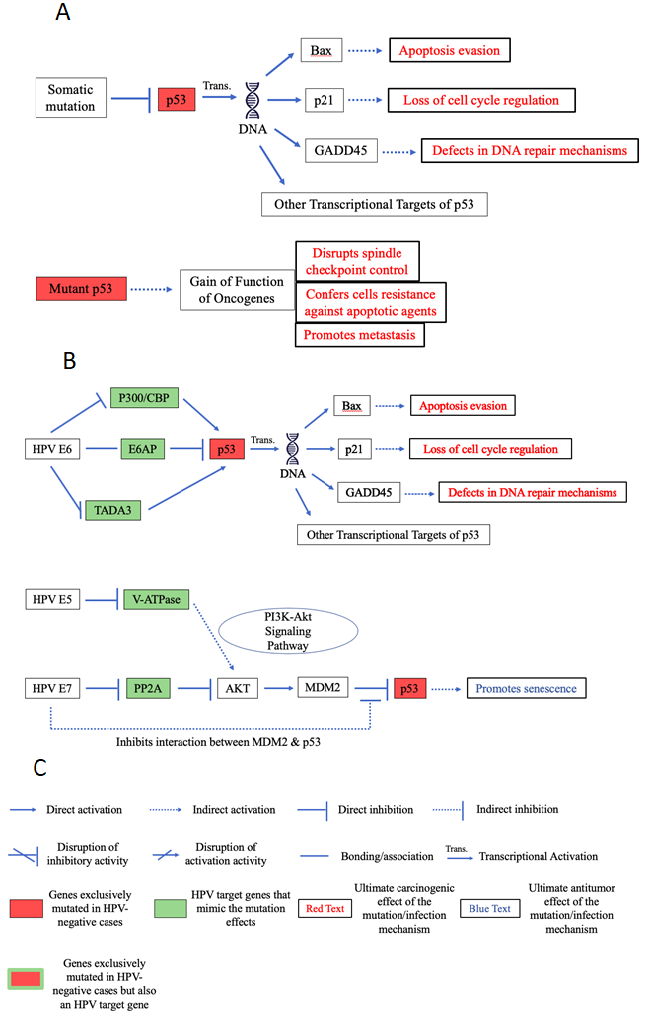
Figure 4: Pathway diagrams of p53 carcinogenic effects through HPV and mutation.
A. Pathway diagram of p53 mutation-induced carcinogenic effects. B. Pathway diagram of HPV-cancer-supporting effects that focus around p53. While HPV is able to mimic the p53 transcriptional inhibition induced by somatic mutations, it is unable to mimic the gain-of-function of oncogenes that mutations confer, as it doesn’t seem to be able to promote metastasis. Additionally, HPV’s inhibitory effects on p53 is counteracted by the E7 viral protein through E7’s inhibition of MDM2-p53 interaction. C. Key for interpreting the pathway diagram. This key will apply to all the pathway diagrams included in this paper.
With the self-contradicting effects produced by this E7 function that inhibits MDM2–P53 interactions, we speculate that the effects of HPV E6 and E7 on P53 somewhat compensate for each other. As HPV E6 suppresses P53, E7 enhances P53 activity, so the effects of the two viral proteins contradict each other, possibly reducing the amount of repression P53 receives and thus conferring some survival advantage to HPV-positive cancer patients when compared to those HPV-negative, driver-mutation-based cancer patients. This speculation is supported by the fact observed in some studies that the expression of P53 as detected through immunohistochemistry is significantly higher in HPV-positive cancers than in HPV-negative ones for cancers of the upper respiratory tract and in laryngeal tumors49,50. The study results of Gillison and co-authors4, and Maruyama and colleagues51, also support our speculations, as they showed an inverse relationship between HPV-presence and P53 mutation in oropharyngeal carcinomas, with HPV-positive tumors less likely to harbor P53 mutations than HPV-negative ones. However, there have also been experimental findings contradictory to this hypothesis, with Wang and colleagues52 stating that HPV-positive breast cancer tissues showed significantly lower expression of P53 and Gualberto and colleagues39 confirming that result in metastatic cancers of unknown primary in the head-and-neck region. Our speculation also contradicts previous theories that HPV-negative mutation-induced tumor cells accumulate excessive amounts of P53, as the frequently occurring missense mutations may render the P53 transcription factor resistant to proteolytic degradation by E3 ubiquitin ligases such as MDM2, maintaining high levels of the mutant P53 protein53. If such theories were true, we would expect HPV-negative cancers to have higher levels of P53 expression than HPV-positive ones, and we would not see the HPV-positive anticancer effect of E7 on the expression of P53 proposed in our speculation.
We propose that these characteristics—the gain-of-function of oncogenes accompanying P53 somatic mutations and the antitumor effects of E7 and E5 induced by the inhibition of MDM2/P53 interaction—play roles in accounting for the prognosis difference between HPV-positive and HPV-negative tumors.
CDKN2A and CDK6
Somatic mutation of the CDKN2A gene results in functionally deficient p16ink4a and possibly p14arf proteins. Under normal circumstances, the p16ink4a binds to and inhibits CDK4/6, which promote cell cycle progression through the phosphorylation and activation of the retinoblastoma protein (pRb)54, and p14arf interacts with and inhibits P53-degrading MDM2 to lead to the stabilization of P53 and thus cell cycle arrest at the G1/S phase55. With somatic mutation, as demonstrated by Poi with colleagues in head and neck squamous cell carcinomas55, the mutant types of p16 showed significantly lower inhibitory activity than wild type p16, leading to dysregulated CDK4/6 levels and thus to uncontrolled cell cycle progression. On the other hand, functionally deficient mutant p14arf leads to increased levels of MDM2 and thus elevated levels of P53 destruction, further causing loss of cell cycle control55.
In HPV-positive cases, the E7 viral protein has been demonstrated to trigger the overexpression of p16ink4a in both cervical and head and neck squamous cell carcinomas56,57,58. Although in HPV-negative cases, this overexpression would be linked to inhibition of CDK4/6 and thus to antitumor cell cycle arrest, the presence of HPV prevents cell cycle arrest and senescence in these tumors through E7-caused degradation of pRb56,59. This degradation eliminates the tumor-suppressing protein that is essential for the execution of the “arrest signal”59.
In addition to the indirect targeting of p16ink4a described above, HPV infection can bypass the p16ink4a protein and affect downstream targets of this CDKN2A-encoded tumor-suppressing protein. Here we assess the effects of HPV infection on the p16ink4a direct downstream targets CDK4 and CDK6. First of all, as previously mentioned, HPV E5 functionally impairs the V-ATPase enzyme, triggering a series of events in the PI3K-Akt signaling pathway and this time leading to decreased transcriptional activity in producing p27, which is an important factor for cell cycle progression from the G1 to the S phase through their interactions with CDK4/6 (Figure 5C)60. This decreased expression of p27 leaves the remaining p27 unable to inhibit the cell cycle progression factors cyclin D1 and CDK4/6, thus leading to dysregulated cell cycle progression16. As detailed by the KEGG pathway “Human Papillomavirus Infections” and as illustrated in Figure 5C as general HPV infection mechanisms, HPV E5’s activation of PDGFβ receptor (PDGFβR), E7’s inhibition of PP2A, E6’s inhibition of PTEN, the degradation of P53 by the E6/E6AP complex, and the inhibition of p27 by both E5 and E716 all lead to the same consequence of dysregulation of cyclin D1 and CDK4/6. In turn, these viral mechanisms contribute to the dysregulation of cell cycle progression, which is a hallmark of cancer, as the dysregulation can leave DNA damage and mutations uncorrected and thereby generate abnormal tumor cells.
Thus, HPV-infection mechanisms powerfully mimic the effects of functional alterations of p16arf induced through mutations, as the ultimate consequence of cell cycle dysregulation is supported in HPV-positive cases by triggering the PI3K-Akt pathway through seven different target genes. The accumulation of these seven separate triggering events could amplify the signal that leads to dysregulation. Although E7’s triggering to overexpression of p16ink4a functionally contradicts with its pRb-degrading mechanisms, the contradiction’s ultimate effect is pro-cancer, and we found no HPV effects that might decrease its carcinogenic efficiency in our searches.
Investigating how HPV mimics the effects of CDKN2A mutations that alter p14arf, we discovered that the E7 viral protein increases p14arf expression. It cleaves E2F-1/pRb binding, releases active E2F-1, and utilizes the ability of transcriptional factor E2F-1 to positively regulate p14arf and induce p14arf expression. Through this increased p14arf expression, E7 actually inhibits the P53-degrading MDM2, protecting P53 from ubiquitination and thus enhancing the cell cycle arrest mechanisms56,61. Experimental results from Kanao and co-authors56 support the above speculations, as they showed an overexpression of p14arf in all the HPV-positive cervical cancers they studied. Furthermore, as discussed in the TP53 section, the presence of HPV E7 inhibits the interaction between MDM2 and P53, as shown in mouse embryo fibroblasts44. So, even if the p14arf somatic driver mutation was already present before infection and had already induced elevated levels of MDM2, the introduction of HPV inhibits the mutant p14arf’s oncogenic effects, as HPV inhibits the interaction between MDM2 and P53 and protects P53 from degradation. This mechanism, on top of the increased p14arf expression, adds another layer of protection for P53.
So, E7 actually mimics the function of normal, nonmutated p14arf instead of mimicking the effects of p14arf with the somatic mutation. These mechanisms create interesting results, as while E7 protects P53 from degradation, E6 binds to E6AP to promote the degradation of P53, as described in the previous section about P53. This point of viral effects self-contradiction may contribute to the relative inefficiency of HPV infection compared to driver somatic mutations leading to better survivals.
CCND1
The CCND1 gene codes for the cyclin D1 protein, which plays an essential role in the G1 phase cell cycle progression. According to a 2018 study on the expression of CCND1 in endometrial adenocarcinomas62, the somatic mutation of the CCND1 gene occurs most commonly in the C-terminus of CCND1, likely inhibiting GSK3β-mediated phosphorylation and inhibition of cyclin D162. With this inhibition, cyclin D1 levels become elevated, promoting cell cycle progression.
In HPV infection, the exact same set of genes leading to changes in CDK4/6 expression, as discussed above, are affected by the virus to support changes in CCND1. The only difference exists in that in addition to inhibition of P27, inhibition of CCND1 can be repressed through Akt’s inhibition of GSK3 β (see the two separate branches stemming from Akt in Figure 5C). Similar to the CDK4/6 scenario, the inhibition of P27 leads to insufficient repression of cyclin D1 and thereby to dysregulated progression of the cell cycle, which is a significant contributing factor to unchecked cell growth and the development of genetically abnormal tumor cells.
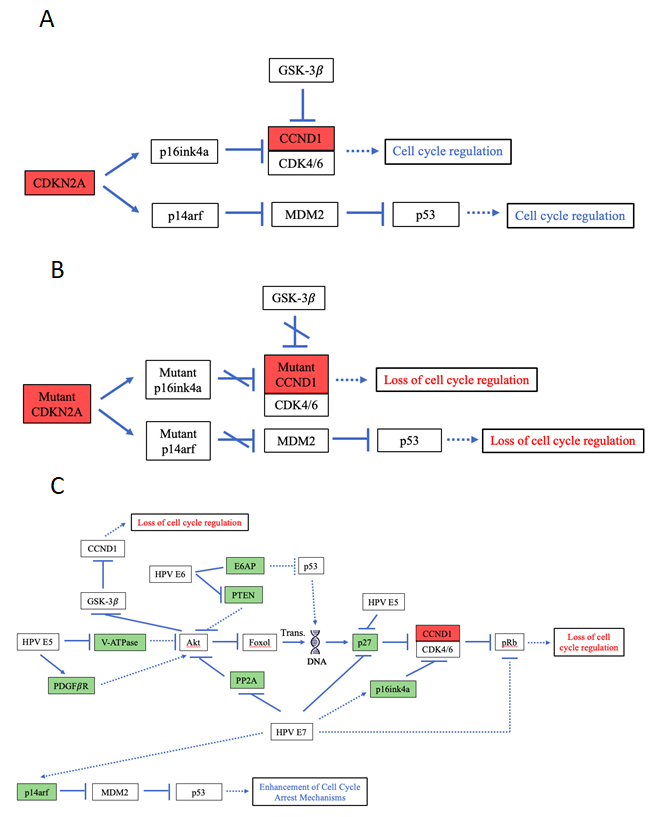
Figure 5: Pathway diagrams for normal functions of and HPV- and mutation-induced effects of CDKN2A & CCND1.
A. Pathway diagram of the functions of wild type CDKN2A, CDK6 and CCND1. B. Pathway diagram of somatic-mutation-induced carcinogenic effects for CDKN2A and CCND1. C. Pathway diagram of HPV-caused effects focusing around CDKN2A, CDK6 and CCND1. As shown in the diagram, HPV affects a set of eight gene targets and powerfully mimics the loss of cell cycle regulation effect induced by the somatic mutations in CDKN2A and CCND1. However, HPV E7’s indirect interaction with p14arf leads to an antitumor effect that enhances cell cycle arrest and somewhat undermines the previous powerful loss of cell cycle regulation effect.
The nearly complete similarity between the way HPV affects the P16INK4a downstream targets, CDK4/6, and the way it affects cyclin D1 is not surprising, as the cyclin D1 protein actually forms a complex with CDK4/6, known as the cyclin D/CDK4/6 complex63.
FGFR1
The fibroblast growth factor receptor 1 (FGFR1), a cytokine receptor, initiates a cascade of intracellular signaling in the PI3K-Akt and the MAPK signaling pathways upon its binding to fibroblast growth factors64, eventually leading to gene transcriptions that are related to cell cycle regulation, migration and cell survival65. FGFR’s “family members” FGFR2 and FGFR3 have been shown to interact with the bovine papillomavirus (BPV) E2 protein to impair viral replication66, but the study by DeSmet and colleagues66 failed to identify any associations between FGFR1 and HPV E2. The FGFR1 mutations or more frequently, amplifications, can cause aberrant activations of its downstream pathways through abnormal phosphorylation and can lead to antiapoptotic and mitogenic effects65. The mutation in this gene has also been shown to induce transformation in the NIH3T3 mouse embryonic fibroblast cell line67.
Interestingly, heparan sulfate proteoglycans, which modulate binding of FGF to its receptor, has been shown to be essential for successful HPV infection68,69. In fact, one of the heparan sulfate proteoglycans, syndecan1, is demonstrated to be involved in HPV penetration to the cell70. With this association, we speculate that the heparan sulfate proteoglycans facilitate easier activation of pathways that lead to cell growth and thus facilitate carcinogenesis, as membrane heparan sulfate proteoglycans can act as coreceptors for a series of tyrosine kinase-type growth factor receptors and lower their activation thresholds71. We didn’t find any other links between FGFR1 and HPV.
IGF1R
IGF1R has long been associated with tumorigenesis and growth72. Activated by its ligands IGF1 and IGF2, it protects cells from apoptosis73. The C-terminal domain of IGF1R mediates protein-to-protein interactions, which in turn play roles in IGF1R downstream signaling to multiple pathways including the PI3K-Akt and the MAPK signaling pathways, leading to IGF1R’s implication in antiapoptotic responses73. Regarding IGF1R mutations, however, studies have shown conflicting results. Some demonstrated that mutations increase IGF1R’s ability to help tumor cells evade apoptosis through either increased binding of a positive mediator of IGF1R signaling, blocked binding of an inhibitory protein72, or deletions of the C-terminus74,75, while others demonstrated a promotion of apoptotic functions through IGF1R C-terminus point mutations76.
While the HPV Infection KEGG pathway16 didn’t indicate any associations between HPV and IGF1R, Steller and co-authors73, through their study using embryonic mouse fibroblasts with disrupted IGF1R to investigate the role of IGF1R in HPV E6 and E7-induced transformation, suggested that HPV E6 may functionally substitute for IGF1R to protect cells from apoptosis, and Pickard with colleagues77 suggested that HPV E6 and E7 can deplete IGFBP2 expression, thus generating signaling through the IGF1R and FGFR2b to IGF1R downstream pathways.
ERBB2 (HER2)
The ERBB2 gene, also known as HER2, is a member of the EGFR family of transmembrane receptor tyrosine kinases and activates signaling pathways regulating cellular proliferation and survival78. Herter-Sprie and co-authors79 summarized three types of somatic mutations of ERBB2: missense mutations in the kinase domain, missense mutations in the extracellular domain and large deletions of the extracellular domain. All three types of somatic mutations activate the ERBB2 protein, leading to downstream signaling in a multitude of pathways including the MAPK and the PI3K-Akt signaling pathway80,81. Such mutational activation and induced downstream signaling lead to proliferation and apoptosis evasion (Figure 6A).
Studies have proposed that HPV viral proteins cooperate with ERBB2 to stimulate transformation of normal oral and cervical epithelium82-84. In experiments with oral epithelial cells, the co-expression of E6/E7 and ERBB2 lead to the downregulation of the E-cadherin/catenin complex, which plays important roles in epithelial cell-to-cell adhesion82. This downregulation causes cell-adhesion molecule loss, which has been causally associated with loss of intercellular adhesion, gain of invasive properties and the development of epithelial cancers85,86, rather than being designated as a consequence of tumor progression. Therefore, the presence of HPV E6/E7 can lead to tumor cell invasion and contribute to metastasis87.
Additionally, the HPV-positive status has been linked to higher levels of ERBB2 expression in HNSCC82,88. This ERBB2 overexpression has been associated with the gain of function of several oncogenic pathways caused by p53 mutants89, which induce genomic instability and metastasis (as mentioned in the p53 section) that HPV-affected p53 doesn’t seem to be able to mimic. Also, we speculate that this overexpression can suggest that HPV affects ERBB2, as both the increased ERBB2 expressions and ERBB2 somatic mutations can lead to downstream signaling in the MAPK and PI3K pathways. However, the specific mechanisms behind such mimicries are not well understood.
HRAS
HRAS is a member of the Ras gene family that plays important roles in the MAPK signaling pathway, which receives external signals that arise from the presence of mitogens and promotes cell growth and proliferation90. The activation of the MAPK pathway is actually triggered by activations of previously mentioned ERBB2, IGF1R, and FGFR1 genes (Figure 6A). Mutations in HRAS maintain the HRAS GTPase in its GTP-bound active state, thus producing permanently activated HRAS proteins and therefore constantly activated MAPK pathway signaling process91,92, which eventually leads to dysregulated cell cycle progression and division that are significant hallmarks of cancer. Somatic missense mutations in the Ras family of genes have also been shown to confer gain-of-function mechanisms to oncogenes93. As previously mentioned, Ras’ cooperation with mutant types of p53 induce metastasis and angiogenesis, as observed in non-small cell lung carcinoma cells42,43.
The HPV E5 viral protein, which inhibits the functions of V-ATPase when studied in transfected human keratinocytes47, seems to be the main mutation-mimicking agent for HRAS. Through the inhibition, E5 leaves V-ATPase unable to inhibit EGFR’s activation of PI3KCA and thus the MAPK signaling pathway. Thus, E5 triggers a cascade of effects that will eventually activate the Ras family of genes, thus indirectly activating the MAPK signaling pathway (Figure 6B). In addition, as shown in the HPV Infection KEGG Pathway16, HPV E5 directly activates PDGFβR, which in turn activates the MAPK signaling pathway through Grb2 (Figure 6B). However, some studies have indicated that interaction with PDGFβR is only a characteristic of BPV94,95, raising doubts about the previously described HPV interaction with PDGFβR.
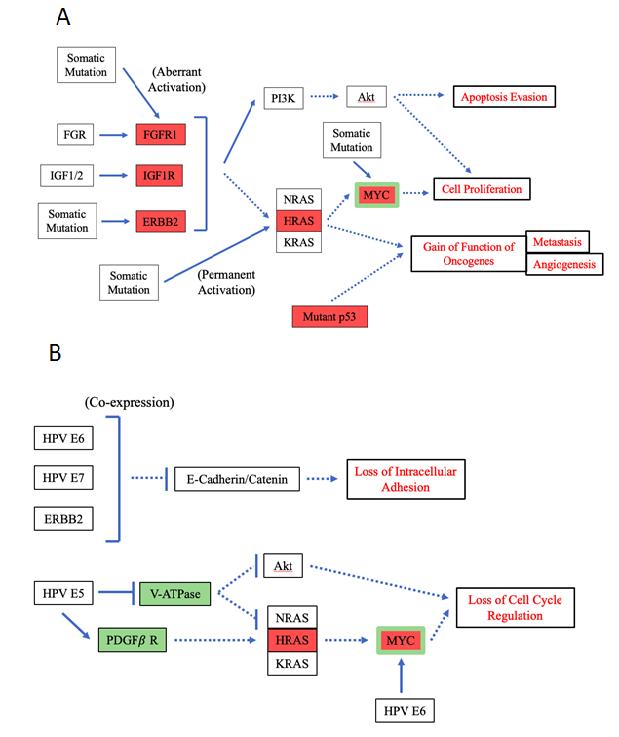
Figure 6: Pathway diagrams for mutation and HPV-induced effects of FGFR1, IGF1R, ERBB2, HRAS & MYC.
Processes (either mutations or HPV infections) for which the effects are still under debate or completely unknown are not included in this diagram. This statement applies to FGFR1 in the HPV infection diagram and to IGF1R in both the mutation and HPV infection diagrams. As the diagrams illustrate, HPV is largely able to mimic the loss of cell cycle regulation effects caused by mutations through the targeting of genes that function in the MAPK and PI3K-Akt pathways. It is also able to support loss of intracellular adhesion and thus promote metastasis, effects not achievable by the mutations. However, it is not unable to mimic the gain-of-function-of-oncogene effects of mutations. A. Pathway diagram for the effects of somatic mutations of FGFR1, IGF1R, ERBB2, HRAS and MYC. B. Pathway diagram for the effects of HPV infection focusing around FGFR1, IGF1R, ERBB2, HRAS and MYC.
MYC
MYC is an important regulator of both cell growth and cell metabolism96, functioning in multiple different pathways. MYC binds to MAX, and the complex’s normal functions involve the transcriptional repression of cell cycle inhibitor-genes such as p15 and p2197, transcriptional activation of pro-proliferative cell-cycle regulating genes such as Cyclin D196 and control of DNA replication98. With these important responsibilities, MYC is normally tightly regulated by mitogenic signals.
In tumor cells, however, the expression of MYC is almost always increased, sometimes by mutations in the gene itself but usually by the induction of its expression by upstream oncogenic alterations, such as HRAS and IGF1R mutations leading to changes in the MAPK signaling pathway96. Such an overexpression causes rapid cell proliferations enabled through the overactivation of pro-proliferative genes, under-inhibition of cell-cycle inhibitor-genes and increased DNA replication activities.
Previously, it has been demonstrated that the integration of HPV viral DNA in genital tumors activated MYC genes99. Other studies100-102 showed that HPV E6 and MYC associate in vitro and in vivo in a complex. Such an interaction enhances MYC activity and thus is a mutation-mimicking mechanism by the virus.
In addition, as mentioned above, changes in MYC are often caused by changes in MYC’s upstream regulators. Therefore, HPV-targeting of MYC’s upstream regulators in the MAPK signaling pathway indirectly affects MYC as well, with E5-induced activation of PDGFβR and E5’s inhibition of V-ATPase eventually leading to the activation of MYC (Figure 6B). These mechanisms all contribute to HPV’s mimicry of mutation leading to MYC overexpression.
KEAP1, NFE2L2 and CUL3
KEAP1-NRF2 (protein encoded by the NFE2L2 gene) interaction is important in protecting cells from endogenous and exogenous stresses. In this interaction, KEAP1 functions as a substrate adaptor protein for the CUL3-containing E3 ubiquitin ligase complex, which mediates NRF2 ubiquitylation103. This ubiquitylation marks NRF2 for degradation and thus maintains a low expression of genes regulated by NRF2 (cytoprotective proteins HO-1, NQO1, TXNRD1 and the glutathione S-transferase (GSTs) (Figure 6A) that help eliminate oxidative stress when needed to do so. At the same time, KEAP1 functions as a sensor for chemical signals induced by oxidative and electrophilic stresses. Upon the detection of these chemical signals, KEAP1 loses the ability to mediate NRF2 ubiquitylation, allowing NRF2 to accumulate and activate the expression of its cytoprotective target genes, which mainly balance the free radicals and antioxidants in the body to ameliorate the oxidative stress problem104,103 (Figure 7A).
A study of KEAP1 and NFE2L2 mutations in non-small cell lung squamous cell carcinomas105 identified that somatic mutations of KEAP1 usually implicate a KEAP1 loss of function, while those of NFE2L2 interrupt binding of NRF2 to KEAP1105. A separate study of CUL3 mutations in papillary renal cell carcinomas identified that the mutations of CUL3 cause a complete CUL3 loss of function through deletion106. Thus, all of these somatic mutations disable the NRF2-degradation mechanisms and thereby increase the level of intracellular NRF2 and the synthesis of antioxidant and detoxification enzymes, which are the cytoprotective targets of NRF2105,106 (Figure 7B). While the increased synthesis of antioxidant and detoxification enzymes aren’t necessarily carcinogenic effects, the effects of these mutations are also linked to resistance to chemotherapy. A past study by Zhang et al.107 demonstrated that downregulation of NRF2 triggered chemotherapy sensitivity, implying that upregulation of NRF2 would induce the opposite effect of chemotherapy resistance (Figure 7B). Despite this effect, Frank and co-authors105 argue that neither NFE2L2 nor Keap1 mutations are drivers, as the majority of non-small-cell lung carcinomas they observed harbored these mutations in co-occurrence with other cancer-related mutations. In fact, it has been shown that oncogenic K-Ras and MYC contribute to the transcriptional induction of Nrf2108.
By mapping a global network of HPV virus-host interactions, Eckhardt with colleagues109 recently showed that in HPV-positive cases, E1 viral protein physically interacts with and binds to KEAP1 to inactivate KEAP1’s functions, thereby leading to freed NRF2 and thus to expression of NRF2’s cytoprotective target genes (Figure 7C). As Eckhardt and co-authors109 pointed out, this E1-KEAP1 interaction mimics the inactivating mutations in the interaction between KEAP1, NRF2 and CUL3, but the effect mimicked isn’t necessarily carcinogenic but can be linked to the resistance against chemotherapy.
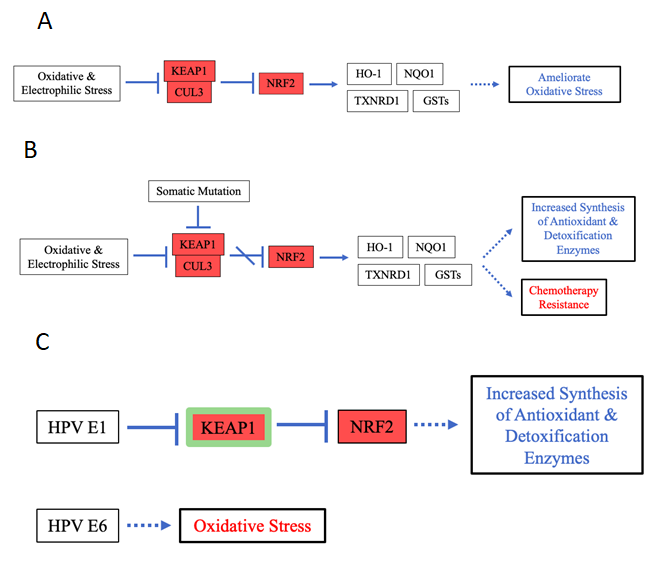
Figure 7: Pathway Diagrams for the normal functions of and mutation- and HPV-induced effects of KEAP1, NFE2L2 and CUL3.
A. Pathway diagram for the normal functions of KEAP1, NFE2L2 and CUL3. B. Pathway diagram for the mutation effects of KEAP1, NFE2L2 and CUL3. C. Pathway diagram for the HPV infection-effects that relate to KEAP1, NFE2L2 and CUL3. Here, HPV infection mimics both the mutation’s disruption of KEAP1–NRF2 interaction and the mutation effect of increased synthesis of antioxidant and detoxification enzymes, however, this effect doesn’t necessarily contribute to carcinogenesis. On the other hand, HPV infection and somatic mutation yield different carcinogenic effects, with HPV inducing oxidative stress and mutation conferring chemotherapy resistance.
In addition, HPV E6 has been shown to support oxidative stress110 (Figure 7C), and the reactive oxygen species released during oxidative stress can chemically damage the DNA111, contributing to virus-induced mutagenesis.
FADD and CASP8
FADD plays an important role in receptor-mediated extrinsic apoptosis, which is triggered by extracellular signals and is to be distinguished from intrinsic apoptosis that is triggered by intracellular signals such as oxidative stress and DNA damage112. In response to extracellular triggers such as killer lymphocytes, Fas is activated (by the Fas ligands carried by those triggers) and binds to FADD via their homologous death domains. This binding triggers the construction of a death-inducing signaling complex (DISC) to eventually lead to the caspase cascade, activating multiple caspases, which then cleave intracellular substrates such as lamin A and PARP to cause apoptosis112. CASP8 is one of these caspases activated (Figure 8A).
A point mutation in FADD inhibits its ability to bind to Fas113, thereby disrupting the apoptosis response to extracellular signals, which now can’t reach the proteins that actually carry out apoptosis. Somatic mutations in CASP8 have been associated with loss of apoptotic functions in a variety of cancers, as CASP8 is rendered nonfunctional114-116. In addition, CASP8 mutations have been shown to confer gain of function mechanisms to other oncogenes in HNSCC, specifically by activating the NF-κB signaling pathway117. Such an activation leads to anti-apoptotic and pro-proliferation effects by NF-κB16,118 (Figure 8B).
During HPV infection, HPV E6 binds to and degrades FADD (Figure 8C), thus protecting cells from apoptosis by preventing transmissions of apoptotic signals through the Fas-FADD pathway and accomplishing similar effects with FADD somatic mutations16,119. However, these similar effects are achieved in slightly different ways, as mutations disrupt FADD’s ability to bind with Fas, while HPV degrades FADD altogether.
Interestingly, Filippova and colleagues120 identified different effects on CASP8 for different isoforms of HPV E6 that arise from alternative splicing120,121. The large E6 isoforms were shown to degrade CASP8, while the small isoforms stabilized CASP8. Thus, the large isoforms inhibited the interaction between FADD and CASP8 and in effect prevented the apoptosis signal from being carried out, while the small isoforms were not able to do so. HPV also seems to mimic the gain-of-function effect that CASP8 mutations confer on the NF-κB pathway, although through a different mechanism. Specifically, HPV E7 inhibits PP2A to increase Akt levels, thus triggering an indirect activation of NF- κB16 (Figure 8C).
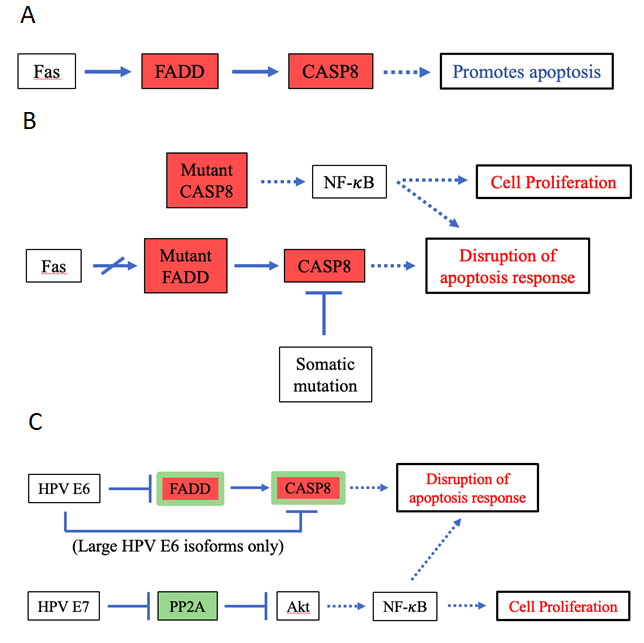
Figure 8: Pathway diagrams for the normal functions of and mutation- and HPV-induced effects of FADD & CASP8.
A. Pathway diagram for the normal functions of FADD and CASP8. B. Pathway diagram illustrating the effects of FADD and CASP8 mutations. C. Pathway diagram illustrating the effects of HPV infection that are related to FADD and CASP8. Although activating NF-κB through slightly different mechanisms than the mutations, HPV powerfully mimics the effects of somatic mutations in FADD and CASP8. However, only large HPV E6 isoforms are able to degrade CASP8, potentially rendering HPV infection as a weaker and less efficient carcinogenic process than the somatic mutations.
BIRC2 (cIAP)
BIRC2 is in the inhibitors of apoptosis protein (IAP) family and inhibits apoptosis through an inactivation of multiple caspases including CASP7 and CASP9, as shown in the Apoptosis KEGG Pathway16, and through its regulatory roles within the NF-κB pathway. The effects of somatic mutations of BIRC2 are still not clear, but these mutations might activate the NF-κB pathway to achieve anti-apoptosis effects in HNSCC and other types of cancers, as suggested by Leemans et al.122.
We didn’t find any interactions between HPV and BIRC2 through our literature searches and pathway analyses. However, we’d like to note that if BIRC2 somatic mutations do activate the NF-κB pathway, a similar carcinogenic effect is already achieved by HPV’s inhibition of PP2A, perhaps eliminating the need for the mimicry of BIRC2 mutation effects.
Discussion
Several perplexing questions surround the topic of HPV-supported carcinogenesis: why is HPV alone incapable of causing cancer, and why do HPV-positive cancers exhibit better prognosis than HPV-negative cancers? To address these questions, in this study, we have presented a concept that HPV infection substitutes for certain driver mutations by mimicking their mutation effects on the pathway to cancer. This mimicry may be accomplished through viral proteins targeting a set of genes that may differ from the genes that are mutated in HPV-negative cancers, but the ultimate effects are similar. Such a model requires HPV infection to be coupled with an appropriate set of mutations in order for progression to cancer to occur, since HPV does not target the full set of cancer driver genes.
We have found ample evidence that support our concept. First of all, we noticed that in HPV-associated cancers such as cervical cancer and head-and-neck cancer, HPV-negative cases generally have more somatic mutations than HPV-positive cases do17,19, and that the majority of mutated genes have a higher mutation frequency in HPV-negative cases19. These data confirm our proposal, as they correspond to the reasoning that since HPV mimics certain somatic mutations, HPV-positive cancers would require the presence of a smaller number of somatic mutations because the presence of HPV supports the carcinogenic effects that originally needed to be induced by somatic mutations. However, we’d like to point out that this mutation frequency difference is subject to intratumor heterogeneity, as the mutation profiles differ to a certain degree across different regions within the same tumor and across metastatic tumor cells. Therefore, the mutation frequencies data we referenced17,19 might not capture the entire mutational landscape and furthermore could be subject to the limitations of the whole-exome sequencing techniques used. In a 2013 study by Zhang et al. investigating the heterogeneity of HNSCC123, the use of whole-genome instead of whole-exome sequencing showed greater HPV-positive HNSCC mutation frequencies than previously observed, revealing a large number of mutations in the non-coding introns. The impacts of these mutations in non-coding sections of the genome are becoming increasingly accepted123 and are very pertinent to our study, as they can confer HPV-positive tumors additional oncogenic potential and could possibly undermine the difference in mutation frequencies we noted based on past studies. The effects of these mutations are worth investigating in the future.
Secondly, we raise an objection to the previous hypothesis that the prognosis difference between HPV-positive and HPV-negative cancers are due to age instead of the presence of HPV, as data from studies conducted by Lei and co-authors14 and Rettig with colleagues15 clearly demonstrate that regardless of age, HPV-positive prognosis rates are better than HPV-negative ones. Thus, establishing HPV as an important prognostic factor, we are able to relate our proposal to the prognosis differences between HPV-positive and HPV-negative cases, as we reason that the differences between mutation and HPV viral mechanisms can possibly account for the prognosis difference.
Finally, in an effort to elucidate how HPV could account for the prognosis difference, we analyzed the differences between the effects of HPV viral infections and those of somatic mutations on 17 specific genes: TP53, CDKN2A, FAT1, CCND1, MYC (c-MYC), NFE2L2, CASP8, FGFR1, CDK6, BIRC2 (cIAP), AJUBA, CUL3, EPHA2, ERBB2, KEAP1, HRAS, and IGF1R. These genes are extracted from The Cancer Genome Atlas mutation frequency database and are the ones that are exclusively mutated in HPV-negative cases. In this analysis, by reviewing past experimental evidence of virus-host interactions and of mutation effects, we have pieced together many ways in which HPV powerfully mimics the effects of somatic mutations. With the exception of EPHA2, AJUBA, BIRC2 and FGFR1, all other extracted genes’ functions were altered in one way or another by HPV to achieve mutation-mimicking effects. In other words, the carcinogenic effects of 13 out of the 17 selected genes are mimicked by HPV. These oncogenic effects of mutations in EPHA2, AJUBA, BIRC2 and FGFR1 that are not mimicked by HPV (to the best of our knowledge at the time of this study) potentially contribute to the prognosis difference conferred by the different HPV statuses, as mutations in HPV-negative cases disrupt cancer pathways that are not as affected in HPV-positive cases. Additionally, it is worth noting that some of the effects are achieved by mutations and by HPV in drastically different ways. For example, while an HRAS mutation directly causes the permanent activation of the MAPK pathway and leads to dysregulated cell cycle progression, HPV E5 accomplishes a similar effect by inhibiting V-ATPase, a far upstream regulator of HRAS. This inhibition would then lead to a cascade of effects that eventually activate the MAPK pathway. Although achieving the similar effect of MAPK-pathway activation, these different methods may confer differences in the degrees of severity of the effect, as direct activations of the pathway by mutations may be more efficient and effective than indirect activations by viruses. As an additional example, mutant p16 leads to uncontrolled cell cycle progression as it demonstrates a reduced inhibition of cell cycle progression promoter CDK4/6 when compared to its wild type counterpart, while HPV triggers the overexpression of p16, which would naturally induce cell cycle arrest, but then prevents cell cycle arrest as it degrades pRb, thereby eliminating the protein that is essential for carrying out the cell cycle arrest. These two methods achieve the similar effect of inducing cell cycle dysregulation, but the degree of severity of the effects might be different. These degree-of-severity differences are not addressed in this current study but should be investigated in the future, as they could be major contributors to the prognosis difference between HPV-positive and HPV-negative cases.
Further addressing the prognosis difference, we have also identified and described multiple HPV infection mechanisms that have been previously uncovered by experimental procedures and that possibly reduce the virus’ efficiency in supporting cancer. First of all, a viral protein sometimes supports effects that undermine the carcinogenic effects of another viral protein. In other words, the virus sometimes has self-contradicting effects. Such a scenario can be seen in HPV E7’s inhibition of interaction between MDM2 and p53, as this inhibition undermines a combined effort of multiple viral proteins to activate MDM2, which degrades p53. Secondly, the virus is unable to support some of the gain-of-function of other oncogene effects that somatic mutations induce. This is seen in the case of TP53, the mutation of which, on top of inducing effects through the inhibition of p53’s transcription mechanisms, increases genomic instability and confers more resistance to a number of antitumor agents. Finally, the differences between various isoforms of viral proteins might introduce another source of “inferiority” for the virus in carcinogenesis. As pointed out in the FADD and CASP8 section, different effects on CASP8 have been identified for different isoforms of HPV E6. Some isoforms confer anti-apoptosis advantages to the tumor cells, while others do not. Such differences between isoforms perhaps render a portion of HPV-supported cancers more dangerous than others. Also, the existence of variants of the same viral proteins due to natural genetic differences raises uncertainty to the consistency of the functions of HPV infection. The same point of argument can be extended to the multiple types of HPVs, including both the high-risk and the low-risk types. More research into the specific differences between isoforms and HPV types would contribute to our overall understanding of the carcinogenic effects of HPV infections.
Overall, our analysis yielded ample evidence supporting our hypothesis for head-and-neck cancers. We expect this hypothesized model to apply to other HPV-associated cancers, such as cervical cancer, because of the model’s ability to account for the differences in the number of somatic mutations in tumors of different HPV statuses, and because of the commonality of this difference across almost all types of HPV-associated cancers.
Conclusion
We proposed a novel explanation for the role of HPV in inducing cancer: the mimicry of the effects of certain somatic mutations. We found ample evidence for this proposal by compiling and analyzing the previously identified viral infection mechanisms that target certain genes to induce carcinogenic effects. This proposal explains the insufficiency of HPV alone to induce cancer, as HPV infections substitute for some, but not all, somatic driver mutations. It also implies the acceleration of the carcinogenic process in HPV-positive cases, and thus we see the trend that HPV-positive cancer patients are younger than HPV-negative ones. Finally, our proposal offers a possible explanation for the better prognosis conferred by HPV-positive cancers, as we showed through compilation of past experimental evidence that the viral mechanisms achieve only partial mimicry of oncogenic mutation effects, that the effectiveness of viral mechanisms differ by the types of HPV, and that certain infection mechanisms contradict other viral carcinogenic effects.
References
- Kobayashi K, Hisamatsu K, Suzui N, et al. A Review of HPV-Related Head and Neck Cancer. Journal of clinical medicine. 2018; 7(9): 241. doi:10.3390/jcm7090241.
- Sarier M, Sepin N, Keles Y, et al. Is There any Association between Urothelial Carcinoma of the Bladder and Human Papillomavirus? A Case-Control Study. Urologia Internationalis. 2019; 104(1–2): 81–86. doi:10.1159/000500467.
- Ni Li , Lin Yang, Yawei Zhang, et al. Human Papillomavirus Infection and Bladder Cancer Risk: A Meta-analysis. The Journal of Infectious Diseases. 2011; 204(2): 217–223. doi:10.1093/infdis/jir248.
- Gillison ML, Koch WM, Capone RB, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000; 92: 709–20.
- Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Sci- ence. 2011; 333(6046): 1157-1160.
- Duenas-González A, Lizano M, Candelaria M, et al. Epigenetics of cervical cancer. An overview and therapeutic perspectives. Molecular cancer. 2005); 4(1): 38.
- Ghittoni R. The role of human papillomaviruses in carcinogenesis. Ecancermedicalscience. 2015; 9. https://doi.org/10.3332/ecancer.2015.526
- Ho GY, Bierman R, Beardsley L, et al. Natural history of cervicovaginal papillomavirus infection in young women. New England Journal of Medicine. 1998; 338(7): 423-428.
- Snijders PJ, Steenbergen RD, Heideman DA, et al. HPVâmediated cervical carcinogenesis: concepts and clinical implications. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland. 2006; 208(2): 152-164.
- de Sanjose S, Brotons M, Pavon MA. The natural history of human papillomavirus infection. Best Practice & Research Clinical Obstetrics & Gynaecology. 2018; 47: 2–13. https://doi.org/10.1016/j.bpobgyn.2017.08.015.
- Bodily J, Laimins LA. Persistence of human papillomavirus infection: keys to malignant progression. Trends in Microbiology. 2011; 19(1): 33–39. https://doi.org/10.1016/j.tim.2010.10.002.
- Chaturvedi AK, Engels EA, Anderson WF, et al. Incidence trends for human papillomavirus related and unrelated oral squamous cell carcinomas in the United States. J Clin oncol 2008; 26: 612-9.
- Mellin H, Friesland S, Lewensohn R, et al. Human papillomavirus (HPV) DNA in tonsillar cancer: clinical correlates, risk of relapse, and survival. International journal of cancer. 2000; 89(3): 300-304.
- Lei J, Ploner A, Lagheden C, et al. High-risk human papillomavirus status and prognosis in invasive cervical cancer: A nationwide cohort study. PLoS medicine. 2018; 15(10): e1002666. doi:10.1371/journal.pmed.1002666.
- Rettig EM, Zaidi M, Faraji F, et al. Oropharyngeal cancer is no longer a disease of younger patients and the prognostic advantage of Human Papillomavirus is attenuated among older patients: Analysis of the National Cancer Database. Oral oncology. 2018; 83: 147–153. doi:10.1016/j.oraloncology.2018.06.013.
- Ogata H, Goto S, Sato K, et al. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic acids research. 1999; 27(1): 29-34.
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015; 517(7536): 576-582. doi:10.1038/nature14129.
- Ma J, Fu Y, Tu YY, et al. Mutation allele frequency threshold does not affect prognostic analysis using next-generation sequencing in oral squamous cell carcinoma. BMC cancer. 2018; 18(1): 758. doi:10.1186/s12885-018-4481-8.
- Banister CE, Liu C, Pirisi L, et al. Identification and characterization of HPV-independent cervical cancers. Oncotarget. 2017; 8(8): 13375–13386. doi:10.18632/oncotarget.14533.
- Iranzo J, Martincorena I, Koonin EV. Cancer-mutation network and the number and specificity of driver mutations. Proceedings of the National Academy of Sciences of the United States of America. 2018; 115(26): 6010-6019.
- Ragin CC, Taioli E. Survival of squamous cell carcinoma of the head and neck in relation to human papillomavirus infection: review and meta-analysis. Int J Cancer. 2007; 121(8): 1813â1820. doi:10.1002/ijc.22851.
- Dayyani F, Etzel CJ, Liu M, et al. Meta-analysis of the impact of human papillomavirus (HPV) on cancer risk and overall survival in head and neck squamous cell carcinomas (HNSCC). Head Neck Oncol. 2010; 2: 15. Published 2010 Jun 29. doi:10.1186/1758-3284-2-15.
- Larsson GL, Helenius G, Andersson S, et al. Human papillomavirus (HPV) and HPV 16–variant distribution in vulvar squamous cell carcinoma in Sweden. International Journal of Gynecologic Cancer. 2012; 22(8): 1413-1419.
- Pintos J, Franco EL, Black MJ, et al. Human papillomavirus and prognoses of patients with cancers of the upper aerodigestive tract. Cancer. 1999; 85: 1903-1909.
- Riethdorf S, Friedrich RE, Ostwald C, et al. p53 gene mutations and HPV infection in primary head and neck squamous cell carcinomas do not correlate with overall survival: a longâterm followâup study. J Oral Pathol Med. 1997; 26: 315–21.
- Snijders PJ, Scholes AG, Hart CA, et al.Prevalence of mucosotropic human papillomaviruses in squamousâcell carcinoma of the head and neck. Int J Cancer. 1996; 66: 464–9.
- Benson E, Li R, Eisele D, et al. The clinical impact of HPV tumor status upon head and neck squamous cell carcinomas. Oral oncology. 2014; 50(6): 565-574.
- Burr AR, Harari PM, Ko HC, et al. HPV impacts survival of stage IVC non-oropharyngeal HNSCC cancer patients. Otorhinolaryngology-head and neck surgery. 2018; 3(1).
- Fakhry C, Westra WH, Li S, et al. Improved survival of patients with human papillomavirus–positive head and neck squamous cell carcinoma in a prospective clinical trial. Journal of the National Cancer Institute. 2008; 100(4): 261-269.
- Camilon PR, Stokes WA, Nguyen SA, et al. The prognostic significance of age in oropharyngeal squamous cell carcinoma. Oral oncology. 2014; 50(5): 431-436.
- Muller PA, Vousden KH. p53 mutations in cancer. Nature Cell Biology. 2013; 15(1): 2-8.
- Gatz SA, Wiesmüller L. p53 in recombination and repair. Cell death and differentiation. 2006; 13(6): 1003.
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009; 137(3): 413-431.
- Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003; 22: 9030-9040.
- Chung-Hsiang Yuan, Maria Filippova, Penelope Duerksen-Hughes. Modulation of Apoptotic Pathways by Human Papillomaviruses: mechanisms and Implications for Therapy. Viruses. 2012; 4(12): 3270-3952.
- Chen J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harbor perspectives in medicine. 2016); 6(3): a026104. doi:10.1101/cshperspect.a026104.
- Zimmermann H, Degenkolbe R, Bernard HU, et al. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. Journal of virology. 1999; 73(8): 6209-6219.
- Kumar A, Zhao Y, Meng G, et al. Human papillomavirus oncoprotein E6 inactivates the transcriptional coactivator human ADA3. Molecular and cellular biology. 2002; 22(16): 5801–5812. doi:10.1128/mcb.22.16.5801-5812.2002.
- Gualberto A, Aldape K, Kozakiewicz K, et al. An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95(9): 5166–5171. doi:10.1073/pnas.95.9.5166.
- Lotem J, Sachs L. A mutant p53 antagonizes the deregulated c-myc-mediated enhancement of apoptosis and decrease in leukemogenicity. Proceedings of the National Academy of Sciences of the United States of America. 1995; 92(21): 9672–9676. doi:10.1073/pnas.92.21.9672.
- Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harbor perspectives in biology. 2010; 2(2): a001107. doi:10.1101/cshperspect.a001107.
- Buganim Y, Solomon H, Rais Y, et al. p53 Regulates the Ras circuit to inhibit the expression of a cancer-related gene signature by various molecular pathways. Cancer research. 2010; 70(6): 2274-2284.
- Solomon H, Brosh R, Buganim Y, et al. Inactivation of the p53 tumor suppressor gene and activation of the Ras oncogene: cooperative events in tumorigenesis. Discovery medicine. 2010; 9(48): 448-454.
- Seavey SE, Holubar M, Saucedo LJ, et al. The E7 oncoprotein of human papillomavirus type 16 stabilizes p53 through a mechanism independent of p19(ARF). Journal of virology. 1999; 73(9): 7590–7598.
- Pim D, Massimi P, Dilworth SM, et al. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene. 2005; 24(53): 7830-8.
- Moll UM, Petrenko O. The MDM2-p53 interaction. Molecular cancer research. 2003; 1(14): 1001-1008.
- Finbow ME, Harrison MA. The vacuolar H+-ATPase: a universal proton pump of eukaryotes. Biomedical Journal. 1997; 324(3): 697-712.
- Long X., Crow MT, Sollott SJ, Oet al. Enhanced expression of p53 and apoptosis induced by blockade of the vacuolar proton ATPase in cardiomyocytes. The Journal of clinical investigation. 1998; 101(6): 1453–1461. doi:10.1172/JCI345.
- Zatonski T, Ciesielska U, Nowinska K, et al. Expression of Cell Cycle-Related Proteins p16, p27, p53 and Ki-67 in HPV-positive and -negative Samples of Papillomas of the Upper Respiratory Tract. Anticancer Journal. 2016; 36: 3917-3924.
- Manjarrez ME, Ocadiz R, Valle L, et al. Detection of human papillomavirus and relevant tumor suppressors and oncoproteins in laryngeal tumors. Clinical cancer research. 2006; 12(23): 6946-6951.
- Maruyama H, Yasui T, Ishikawa-Fujiwara T, et al. Human papillomavirus and p53 mutations in head and neck squamous cell carcinoma among Japanese population. Cancer science. 2014; 105(4): 409–417. doi:10.1111/cas.12369.
- Wang YW, Zhang K, Zhao S, et al. HPV Status and Its Correlation with BCL2, p21, p53, Rb, and Survivin Expression in Breast Cancer in a Chinese Population. BioMed research international. 2017; 2017: 6315v 392. doi:10.1155/2017/6315392.
- Frum RA, Grossman SR. Mechanisms of mutant p53 stabilization in cancer. In Mutant p53 and MDM2 in Cancer. Springer, Dordecht. 2014; 187-197.
- Ohtani N, Yamakoshi K, Takahashi A, et al. The p16INK4a-RB pathway: molecular link between cellular senescence and tumor suppression. The Journal of Medical Investigation. 2004; 51(3, 4): 146-153.
- Poi MJ, Yen T, Li J, et al. Somatic INK4aâARF locus mutations: A significant mechanism of gene inactivation in squamous cell carcinomas of the head and neck. Molecular Carcinogenesis: Published in cooperation with the University of Texas MD Anderson Cancer Center. 2001; 30(1): 26-36.
- Kanao H, Enomoto T, Ueda Y, et al. Correlation between p14ARF/p16INK4A expression and HPV infection in uterine cervical cancer. Cancer letters. 2004; 213(1): 31-37.
- Sano T, Oyama T, Kashiwabara K, et al. Expression status of p16 protein is associated with human papillomavirus oncogenic potential in cervical and genital lesions. The American journal of pathology. 1998; 153(6): 1741-1748.
- Stephen JK, Divine G, Chen KM, et al. Significance of p16 in Site-specific HPV Positive and HPV Negative Head and Neck Squamous Cell Carcinoma. Cancer Clin Oncol. 2013; 2(1): 51â61. doi:10.5539/cco.v2n1p51.
- Munger K, Gwin TK, McLaughlin-Drubin M. p16 in HPV-associated cancers. Oncotarget. 2013; 4(11): 1864.
- Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nature Reviews Cancer. 2010; 10(8): 550.
- Bates S, Phillips AC, Clark PA, et al. p14 ARF links the tumour suppressors RB and p53. Nature. 1998; 395(6698): 124.
- Xu J, Lin DI. Oncogenic c-terminal cyclin D1 (CCND1) mutations are enriched in endometrioid endometrial adenocarcinomas. PloS one. 2018; 13(7): e0199688.
- Dong P, Zhang C, Parker BT, et al. Cyclin D/CDK4/6 activity controls G1 length in mammalian cells. PloS one. (018; 13(1): e0185637.
- Helsten T, Elkin S, Arthur E, et al. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clinical cancer research. 2016; 22(1): 259-267.
- Dienstmann R, Rodon J, Prat A, et al. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Annals of Oncology. March 2014; 25(3): 552–563.
- DeSmet M, Kanginakudru S, Jose L, et al. Papillomavirus E2 protein is regulated by specific fibroblast growth factor receptors. Virology. 2018; 521: 62–68. doi:10.1016/j.virol.2018.05.013.
- Ronchetti D, Greco A, Compasso S, et al. Deregulated FGFR3 mutants in multiple myeloma cell lines with t (4; 14): comparative analysis of Y373C, K650E and the novel G384D mutations. Oncogene. 2001; 20(27): 3553.
- Giroglou T, Florin L, Scha Ìfer F, et al. Human papillomavirus infection requires cell surface heparan sulfate. J Virol. 2001; 75(3): 1565–1570.
- Kumar A, Jacob T, Abban CY, et al. Intermediate heparan sulfate binding during HPV 16 infection in HaCaTs. Am J Ther. 2014; 21(5): 331–342.
- Shafti-Keramat S, Handisurya A, Kriehuber E, et al. Different heparan sulfate proteoglycans serve as cellular receptors for human papillomaviruses. J Virol. 2003; 77(24): 13125–13135.
- Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harbor perspectives in biology. 2011; 3(7): a004952. doi:10.1101/cshperspect.a004952.
- Craddock BP, Miller WT. Effects of somatic mutations in the C-terminus of insulin-like growth factor 1 receptor on activity and signaling. Journal of signal transduction. 2012.
- Steller MA, Zou Z, Schiller JT, et al. Transformation by human papillomavirus 16 E6 and E7: role of the insulin-like growth factor 1 receptor. Cancer research. 1996; 56(21): 5087-5091.
- Liu Y, Lehar S, Corvi C, et al. Expression of the insulin-like growth factor I receptor C terminus as a myristylated protein leads to induction of apoptosis in tumor cells. Cancer research. 1998; 58(3): 570-576.
- Hongo A, Yumet G, Resnicoff M, et al. Inhibition of tumorigenesis and induction of apoptosis in human tumor cells by the stable expression of a myristylated COOH terminus of the insulin-like growth factor I receptor. Cancer research. 1998; 58(11): 2477-2484.
- O'Connor R, Kauffmann-Zeh A, Liu Y, et al. Identification of domains of the insulin-like growth factor I receptor that are required for protection from apoptosis. Molecular and Cellular Biology. 1997; 17(1): 427-435.
- Pickard A, McDade SS, McFarland M, et al. HPV16 Down-Regulates the Insulin-Like Growth Factor Binding Protein 2 to Promote Epithelial Invasion in Organotypic Cultures. PLoS pathogens. 2015; 11(6): e1004988. doi:10.1371/journal.ppat.1004988.
- Connell CM, Doherty GJ. Activating HER2 mutations as emerging targets in multiple solid cancers. ESMO open. 2017; 2(5): e000279.
- Herter-Sprie GS, Greulich H, Wong KK. Activating mutations in ERBB2 and their impact on diagnostics and treatment. Frontiers in oncology. 2013; 3: 86.
- Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007; 26(45): 6469.
- Prenzel N, Fischer OM, Streit S, et al. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocrine-related cancer. 2001; 8(1): 11-31.
- Pollock NI, Wang L, Wallweber G, et al. Increased expression of HER2, HER3, and HER2: HER3 heterodimers in HPV-positive HNSCC using a novel proximity-based assay: implications for targeted therapies. Clinical Cancer Research. 2015; 21(20): 4597-4606.
- Al Moustafa AE, Foulkes WD, Benlimame N, et al. E6/E7 proteins of HPV type 16 and ErbB-2 cooperate to induce neoplastic transformation of primary normal oral epithelial cells. Oncogene. 2004; 23(2): 350.
- Moustafa A, Kassab A, Darnel A, et al. High-risk HPV/ErbB-2 interaction on E-cadherin/catenin regulation in human carcinogenesis. Current pharmaceutical design. 2008; 14(22): 2159-2172.
- Cavallaro U, Christofori G. Cell adhesion in tumor invasion and metastasis: loss of the glue is not enough. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. 2001; 1552(1): 39-45.
- Christofori G, Semb H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends in biochemical sciences. 1999; 24(2): 73-76.
- Martin TA, Ye L, Sanders AJ, et al. Cancer invasion and metastasis: molecular and cellular perspective. In Madame Curie Bioscience Database [Internet]. Landes Bioscience. 2013.
- Mazibrada J, Longo L, Vatrano S, et al. Differential expression of HER2, STAT3, SOX2, IFI16 and cell cycle markers during HPV-related head and neck carcinogenesis. 2014.
- Roman-Rosales AA, Garcia-Villa E, Herrera LA, et al. Mutant p53 gain of function induces HER2 over-expression in cáncer cells. BMC cancer. 2018; 18(1): 709.
- Orton RJ, Sturm OE, Vyshemirsky V, et al. Computational modelling of the receptor-tyrosine-kinase-activated MAPK pathway. Biochemical Journal. 2005; 392(2): 249-261.
- Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer research. 2012; 72(10): 2457-2467.
- Flex E, Jaiswal M, Pantaleoni F, et al. Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Human molecular genetics. 2014; 23(16): 4315-4327.
- Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016; 129(7): 1287-1292.
- DiMaio D, Petti LM. The E5 proteins. Virology. 2013; 445(1-2): 99-114.
- Venuti A, Paolini F, Nasir L, et al. Papillomavirus E5: the smallest oncoprotein with many functions. Molecular cancer. 2011; 10(1): 140.
- Miller DM, Thomas SD, Islam A, et al. c-Myc and cancer metabolism. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012; 18(20): 5546–5553. doi:10.1158/1078-0432.CCR-12-0977
- Gartel AL, Shchors K. Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Experimental cell research. 2003; 283(1): 17-21.
- Dominguez-Sola D, Ying CY, Grandori C, et al. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007; 448(7152): 445.
- Couturier J, Sastre-Garau X, Schneider-Maunoury S, et al. Integration of papillomavirus DNA near myc genes in genital carcinomas and its consequences for proto-oncogene expression. Journal of virology. 1991; 65(8): 4534-4538.
- Gewin L, Galloway DA. E box-dependent activation of telomerase by human papillomavirus type 16 E6 does not require induction of c-myc. Journal of virology. 2001; 75(15): 7198-7201.
- Veldman T, Liu X, Yuan H, et al. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proceedings of the National Academy of Sciences. 2003; 100(14): 8211-8216.
- Zhang Y, Dakic A, Chen R, et al. Direct HPV E6/Myc interactions induce histone modifications, Pol II phosphorylation, and hTERT promoter activation. Oncotarget. 2017; 8(56): 96323.
- Kerins MJ, Ooi A. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Scientific reports. 2018; 8(1): 12846.
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007; 47: 89-116.
- Frank R, Scheffler M, Merkelbach-Bruse S, et al. Clinical and Pathological Characteristics of KEAP1-and NFE2L2-Mutated Non-Small Cell Lung Carcinoma (NSCLC). Clinical Cancer Research. 2018; 24(13): 3087-3096.
- Ooi A, Dykema K, Ansari A, et al. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell carcinoma. Cancer research. 2013; 73(7): 2044-2051.
- Zhang P, Singh A, Yegnasubramanian S, et al. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Molecular cancer therapeutics. 2010; 9(2): 336-346.
- DeNicola GM, Karreth FA, Humpton TJ, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011; 475(7354): 106.
- Eckhardt M, Zhang W, Gross AM, et al. Multiple routes to oncogenesis are promoted by the human papillomavirus-host protein network. Cancer discovery. 2018; 8(11): 1474-1489.
- Williams VM, Filippova M, Filippov V, et al. Human papillomavirus type 16 E6* induces oxidative stress and DNA damage. Journal of virology. 2014; 88(12): 6751-6761.
- Kawanishi S, Hiraku Y, Oikawa S. Mechanism of guanine-specific DNA damage by oxidative stress and its role in carcinogenesis and aging. Mutation Research/Reviews in Mutation Research. 2001; 488(1): 65-76.
- Lee EW, Seo JH, Jeong MH, et al. The roles of FADD in extrinsic apoptosis and necroptosis. BMB reports. 2012; 45(9): 496-508.
- Chinnaiyan AM, O'Rourke K, Tewari M, et al. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995; 81(4): 505-512.
- Soung YH, Lee JW, Kim SY, et al. Caspase-8 gene is frequently inactivated by the frameshift somatic mutation 1225_1226delTG in hepatocellular carcinomas. Oncogene. 2005; 24(1): 141.
- Kim HS, Lee JW, Soung YH, et al. Inactivating mutations of caspase-8 gene in colorectal carcinomas. Gastroenterology. 2003; 125(3): 708-715.
- Soung YH, Lee JW, Kim SY, et al. CASPASE-8 gene is inactivated by somatic mutations in gastric carcinomas. Cancer Research. 2005; 65(3): 815-821.
- Ando M, Kawazu M, Ueno T, et al. Cancerâassociated missense mutations of caspaseâ8 activate nuclear factorâκB signaling. Cancer science. 2013; 104(8): 1002-1008.
- Chen F, Castranova V, Shi X. New insights into the role of nuclear factor-kappaB in cell growth regulation. The American journal of pathology. 2001; 159(2): 387–397. doi:10.1016/s0002-9440(10)61708-7.
- Filippova M, Parkhurst L, Duerksen-Hughes PJ. The human papillomavirus 16 E6 protein binds to Fas-associated death domain and protects cells from Fas-triggered apoptosis. Journal of Biological Chemistry. 2004; 279(24): 25729-25744.
- Filippova M, Johnson MM, Bautista M, et al. The large and small isoforms of human papillomavirus type 16 E6 bind to and differentially affect procaspase 8 stability and activity. Journal of virology. 2007; 81(8): 4116-4129.
- Olmedo-Nieva L, Munoz-Bello JO, Contreras-Paredes A, et al. The Role of E6 Spliced Isoforms (E6*) in Human Papillomavirus-Induced Carcinogenesis. Viruses. 2018; 10(1): 45. doi:10.3390/v10010045.
- Leemans CR, Snijders PJ, Brakenhoff RH. The molecular landscape of head and neck cancer. Nature Reviews Cancer. 2018; 18(5): 269.
- Zhang XC, Xu C, Mitchell RM, et al. Tumor Evolution and Intratumor Heterogeneity of an Oropharyngeal Squamous Cell Carcinoma Revealed by Whole-Genome Sequencing. Neoplasia. 2013; 15(12): 1371–IN7. Https://doi.org/10.1593/neo.131400.