
Rifamycin SV Anti-inflammatory and Immunomodulatory Activities for Treatment of Mucosal and Liver Inflammation
Caridad Rosette, Alessandro Mazzetti, Roberto Camerini, Luigi Moro, Mara Gerloni*
Cosmo Pharmaceuticals, Riverside II. Sir John Rogerson’s Quay, Dublin, Ireland
Abstract
Rifamycin SV (rifamycin), is a member of the ansamycin family of antimicrobial compounds which kills bacteria commonly associated with infectious diarrhea and other enteric infections. For colonic diseases like diverticulitis, inflammatory bowel syndrome (IBS) or inflammatory bowel disease (IBD), bacterial proliferation or microbial dysbiosis is associated with a strong inflammatory component. This inflammation has a profound influence on the liver via the gut-liver axis. This review summarizes the anti-inflammatory activities of rifamycin based on analyses of its impact on two key regulators of inflammation: PXR and NFκB. Rifamycin was found to activate PXR and two of its downstream targets, CYP3A4 and PgP, in liver and intestinal cell lines. Rifamycin also directly inhibited NFκB in a cell line which lacks PXR expression. These dual activities likely explain the inhibition of pro-inflammatory cytokine secretion from human colonic cells lines and activated CD4+ T cells. These experimental data define the immune regulatory characteristics of rifamycin and an emerging role in the treatment of both gastrointestinal (GI) and liver disorders.
Introduction
The gut barrier consists of three interacting components: the mucus layer, the intestinal epithelium, and the mucosal immune system1. The evidence for the role of inflammation and immune regulation in the pathophysiology of GI diseases, such as IBD and IBS is indisputable while their role in diverticular disease is compelling. Inappropriate adaptive immune response leads to infiltration of immune cells in the intestinal mucosa and elevated levels of pro-inflammatory cytokines that amplify and maintain the inflammatory response to intestinal pathogens. In conjunction with this altered immune milieu, changes in gut microbiota and intestinal barrier breakdown allow cytokines and bacterial components or metabolites to reach the portal and systemic circulation. Thus, through the gut-liver network, disruption of intestinal epithelial homeostasis can lead to hepatic exposure to exogenous and endogenous antigens that drives the liver pathology associated with several liver diseases including alcoholic and nonalcoholic fatty liver disease (NALFD) and steatohepatitis2.
PXR is a transcription factor with a well-established role as a xenobiotic sensor to promote xenobiotic metabolism and excretion in the gut and liver, as well as homeostasis of endobiotics such as glucose, lipids, steroids and bile acids. In recent years, however, an increasing number of new roles for PXR have been described. These include modulating atherosclerosis development, vascular functions, as well as inflammation3. These newly discovered functions of PXR clearly indicate that PXR is more than a xenobiotic/endobiotic receptor but represents a central node that links the gut-liver axis by maintaining the gut barrier function and regulates inflammatory mechanisms in both organs4-10. For example, PXR stimulation by bacterial metabolites protects the intestinal barrier by inducing expression of cell junction-associated molecules in intestinal epithelial cells (IECs)6, 11 while PXR stimulation by pregnenolone downregulates lipopolysaccharide (LPS)-induced inflammatory cytokine expression in hepatocytes10.
It has long been shown that inflammation and pro-inflammatory cytokines suppress the expression of drug metabolizing enzymes and transporters in inflammatory disease conditions like IBD, sepsis, and inflammation-induced cholestasis12. The underlying molecular mechanism for this suppression may be due to the reciprocal crosstalk between PXR and another major regulator of inflammation, NFκB13-16. Activated PXR reduces the activity of NFκB17 while activated NFκB reduces the expression and activity of PXR13, 18. PXR and NFκB proteins are abundantly expressed in the liver and in tissues along the GI tract19, 20. However, patients with IBD have significantly lower expression of PXR21 but higher expression of NFκB compared to non-IBD patients22 and abnormal activation of NFκB leads to excessive production of pro-inflammatory cytokines that cause chronic inflammation in the bowel. Likewise, many patients with NALFD or advanced liver disease display increased hepatic NFκB activation19, 23. NFκB signaling plays a crucial role in liver inflammatory responses by controlling the expression of an array of growth factors and cytokines such as tumor necrosis factor-alpha (TNFα) and interleukin 6 (IL6)24.
The antibiotic rifamycin was shown to activate PXR and inhibit NFκB in vitro25. A member of the ansamycin family of antimicrobial compounds, rifamycin is a rifampin structural analog that exerts potent antimicrobial activity against gram-positive, gram-negative, and both aerobic and anaerobic bacteria26. In addition, rifamycin as well as rifaximin have potential roles in the treatment of Clostridium difficile infections27, 28. An extended colonic release formulation, Rifamycin SV MMX®, has been approved in the US and Europe for treatment of traveler’s diarrhea (TD)29. Clinically relevant drug interactions are not expected because of its poor GI absorption leading to low systemic blood levels30. In vitro studies showed that rifamycin activates PXR and its downstream targets, CYP3A4 and PgP25. In parallel, rifamycin was shown to inhibit NFκB transcriptional activity even in the absence of PXR. Importantly and consistent with its influence on NFκB activity, rifamycin showed potent downregulation of inflammatory cytokine secretion (IL2, IL6, IL8, IL17A, IFNγ, and TNFα) from human colonic epithelial cells as well as from cells of the human immune system (CD4 T cells), thereby defining in vitro anti-inflammatory characteristics of rifamycin31. Given these results, this narrative review explores the potential mechanisms of action by which rifamycin may exert a direct therapeutic effect in GI and liver diseases as well as indirectly in liver diseases, via the gut-liver axis.
Rifamycin is a Potent Stimulator of PXR, CYP3A4 Activities and PgP Expression
PXR regulates drug clearance in the liver and intestine via induction of genes involved in drug and xenobiotic metabolism. Stimulation of PXR transcriptional activity induces metabolic enzymes like CYP3A4, which oxidizes xenobiotics like bacterial metabolic products, environmental toxins or drugs, for clearance from the body. Stimulation of PXR also induces expression of xenobiotic transporters like PgP in the intestine and liver3. To evaluate the effect of rifamycin on PXR transcriptional activity, rifamycin and the control antibiotic rifampicin were tested in a HepG2 liver cell line modified to overexpress full length PXR. Rifamycin effectively activated PXR activity, although rifampicin was 3 times more efficacious than rifamycin (EC50= 2.87 x 10-6 M vs. 9.29 x 10-6 M, respectively) (Figure 1A). In these cells, rifamycin also induced high levels of one of PXR’s downstream effectors, CYP3A4. Consistent with rifampicin’s higher PXR activity, the induction of CYP3A4 protein metabolic activity by rifamycin was lower than rifampicin’s (EC50 6.58 x 10-6 vs EC50 1.67 x 10-6 M) (Figure 1B). Thus, within this cell system, the rank order of PXR and Cyp3A4 activation is maintained.
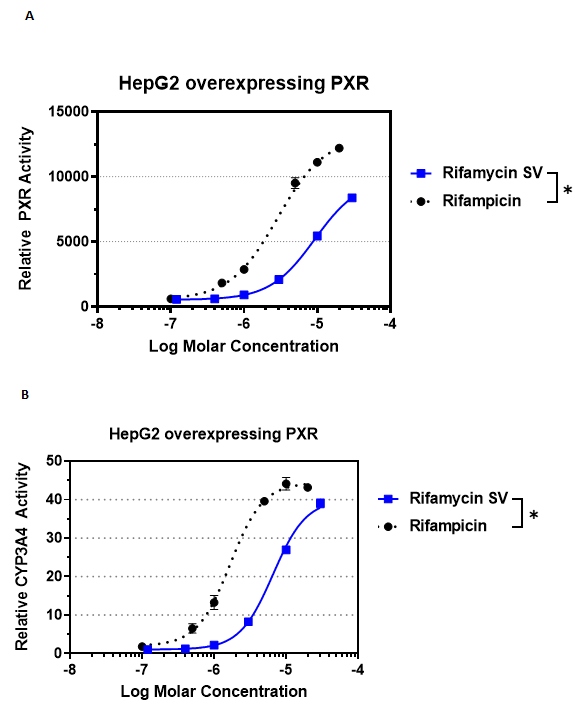
Figure 1: Rifamycin stimulates PXR transcriptional activity and CYP3A4 enzymatic activity.
A) PXR transcriptional activity in HepG2 liver cell line stably overexpressing full length human PXR and luciferase reporter gene linked to promoters identified in the human CYP3A4 gene. Cells were treated for 24 h followed by quantitation of reporter-dependent luminescence. B) Cyp3A4 enzymatic activity was analyzed in the same HepG2 cells overexpressing PXR. Cells were treated with rifamycin or rifampicin for 48 h followed by quantitation of Cyp3A4 conversion of a pro-luciferin substrate. Data points are MEAN + SEM, luciferase units relative to 0.1% DMSO, n = 3 experiments each in triplicate wells. Data were fitted using log agonist versus normalized response, variable slope, 4 parameter curve settings. Data were plotted in GraphPad Prism software (version 7.03, San Diego, CA). EC50 values were obtained from fitted curves. Background values were subtracted from all readings. Reproducibility between experiments was calculated by p value determination using paired 2-tailed t-test or ANOVA with Bonferroni multiple comparisons test using Prism software. (*P < 0.05 rifamycin vs. rifampicin EC50).
To clarify the mechanism of the increased PXR activation by rifamycin, PXR mRNA expression was quantified in HepG2 cells treated with 20 µM rifamycin or rifampicin (Figure 2A). The data indicate that rifamycin increased PXR mRNA expression 2.5-fold relative to the DMSO control while rifampicin did so by 1.5-fold. Despite the marginal induction of PXR mRNA by rifampicin, this control antibiotic induced Cyp3A4 mRNA 4-fold relative to DMSO, suggesting that potent stimulation of PXR transcriptional activity in these cells may be due to post-translational modification of PXR rather than positive feedback regulation via transcriptional induction of PXR mRNA32. Increased CYP3A4 mRNA is consistent with high levels of CYP3A4 metabolic activity in the HepG2 cells overexpressing PXR (Figure 1B).
Another downstream target of PXR, PgP, was analyzed in HepG2 as well as Caco2 colonic epithelial cells (Figure 2B, 2C). Induction of PgP protein expression in HepG2 and Caco2 cells was statistically equivalent with both rifamycin and rifampicin.
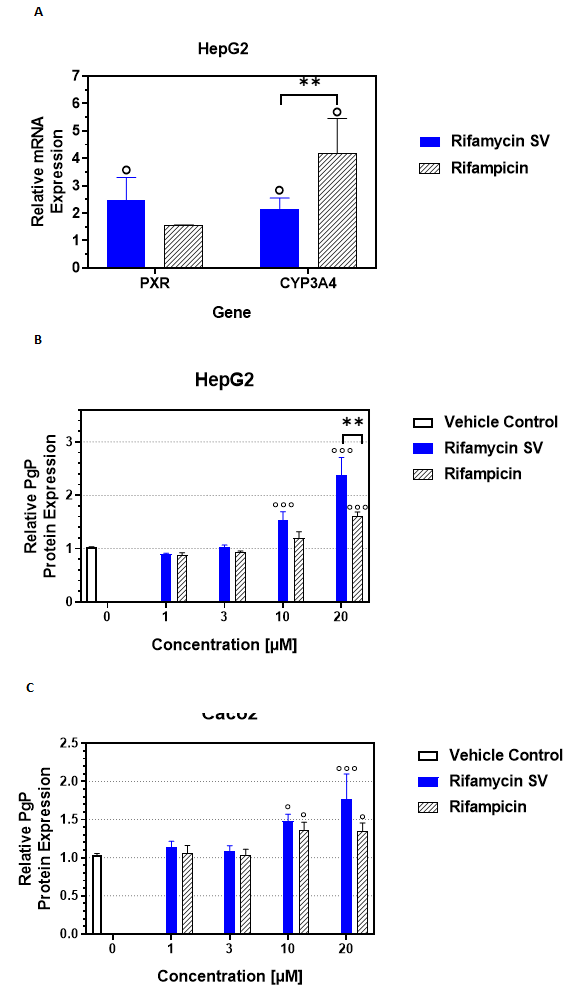
Figure 2: Rifamycin stimulates PXR/CYP3A4 mRNA and PgP protein expression
A) PXR and Cyp3A4 mRNA expression were quantified by qRT-PCR of HepG2 cells treated with 20 µM rifamycin or rifampicin for 4 days. The mean fold change in mRNA expression + SEM relative to 0.1% DMSO are plotted. Experiment was repeated 3 times in triplicates (°p < 0.05 vs. DMSO, **p < 0.005 Cyp3A4 mRNA between rifamycin and rifampicin). PgP protein expression was quantified by flow cytometry in B) HepG2 and C) Caco2 cells treated with rifamycin, rifampicin or DMSO (vehicle control) for 4 days. Data points are MEAN + SEM, relative to DMSO of the mean fluorescence intensity associated with anti-PgP fluorescence, proportional to the PgP protein expression (°P < 0.05, °°°P < 0.0005 vs. DMSO, **p < 0.005 PgP protein between rifamycin and rifampicin at 20 µM in HepG2).
In summary, rifamycin exhibits strong in vitro PXR activation with concomitant CYP3A4 and PgP induction in liver cells. While only PgP protein expression was analyzed in colonic cells due to technical constraints of low PXR and CYP3A4 expression in these cells, the established fact that PXR is a key regulator of CYP3A4 and PgP gene expression in liver and intestinal cells implies that PXR may also be a target of rifamycin in colonic cells33, 34. Thus, these results show that rifamycin can potentially modulate disease-causing crosstalk between the gut and liver.
Rifamycin Inhibits NFκB Transcriptional Activity in a Reporter Cell Line
NFκB is the primary transcription factor that regulates gene expression of pro-inflammatory cytokines and chemokines..To establish if rifamycin inhibits NFκB, a reporter cell line expressing full length NFκB and harboring luciferase reporter gene functionally linked to upstream NFκB genetic response elements, was stimulated with either TNFα or LPS to induce NFκB activity. Rifamycin or rifampicin were dose-titrated onto these inflammatory agonist stimulated cells. Rifamycin antagonized very efficiently both TNFα-induced NFκB activity with IC50 value of 1.48 x 10-5 M (Figure 3A) and LPS-induced NFκB activity with IC50 value of 3.49 x 10-6 M (Figure 3B). In stark contrast, rifampicin did not inhibit TNFα-induced and only weakly inhibited LPS-induced NFκB activities. Since these reporter cells do not express PXR, these results establish that rifamycin is able to inhibit NFκB independently of PXR. In many published cases, activation of PXR leads to inhibition of NFκB transcriptional activity. However, in an acute case of liver inflammation, PXR and NFκB act independently of the other35. Since the NFκB signaling cascade is well recognized as the main pathway activated during the early response to endotoxin36 the direct effect of rifamycin on NFκB, independent of PXR, would also be beneficial in acute cases of endotoxin induced mucosal inflammation.
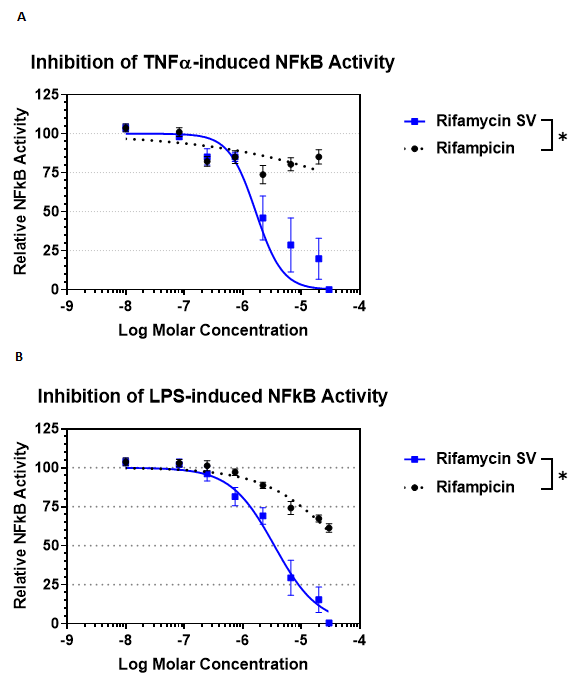
Figure 3: Rifamycin inhibits TNFα- and LPS-induced NFκB transcriptional activity in NFκB reporter cell line.
Inhibition of NFκB transcriptional activity was evaluated in HEK293t reporter cells expressing NFκB and an NFκB-dependent reporter, but not expressing PXR. Cells were treated with A) 30 ng/mL TNFα or B) 100 µg/mL LPS for 24 hr. Data points are %MEAN + SEM, relative to 0.1% DMSO (100% on y-axis), Experiments were repeated 3 times in triplicates (P < 0.05 rifamycin vs. rifampicin IC50).
Interestingly, these data showed that inhibition of NFκB by rifamycin is not dependent on the stimulus since rifamycin potently inhibited induction of NFκB by three distinct stimuli: TNFα, LPS, and PMA (not shown). PMA or diacyl glycerol-activated PKC is important for T cell receptor or IgE-induced NFκB that occurs in immune cells, like T cells and mast cells, respectively37, suggesting that rifamycin would also be effective in directly downregulating inflammation mediated by immune cells.
Rifamycin Inhibits Synthesis of Pro-inflammatory Cytokines
To evaluate if rifamycin can antagonize directly inflammatory cytokine production in cells that are relevant to the pathogenesis of GI diseases, monocytic and macrophage-differentiated THP1 cell lines, and colonic cell lines, HT-29 and Caco2 cells, were induced in vitro to secrete inflammatory cytokines (Figure 4A). Monocytes and macrophages were stimulated with LPS while HT-29 and Caco2 cells were stimulated with IL1ß, in the presence of rifamycin or DMSO. Due to the differential sensitivities of each cell line, rifamycin concentration range for monocytes and macrophages were 50, 100, 150 µM; for HT-29 was 100, 200, 400 µM, while for Caco2 it was 3, 10, 20 µM. At the maximum non-cytotoxic concentrations of rifamycin, secretion of TNFα from monocytes was inhibited by 37%, IL8 from macrophages, HT-29 and Caco2 cells by 65%, 85% and 60%, respectively, compared to the DMSO control. These results clearly show that rifamycin modulates the production of inflammatory cytokines that have been reported to be overexpressed in inflamed colonic mucosa of GI patients.
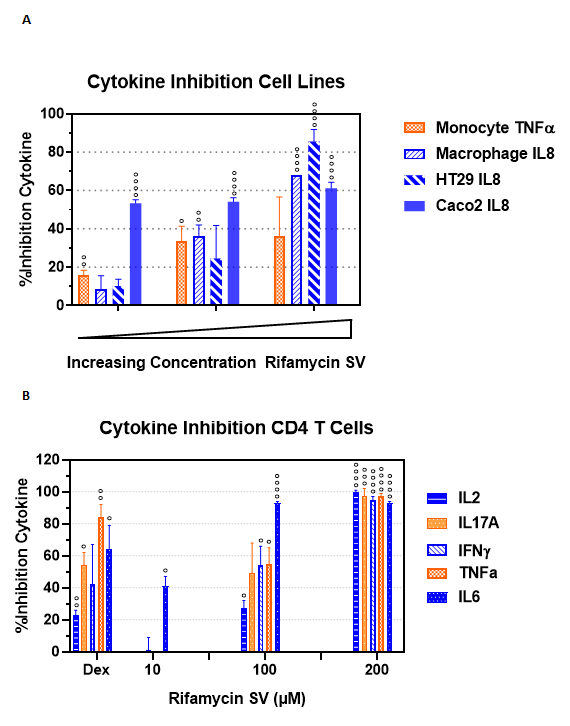
Figure 4: Rifamycin inhibits pro-inflammatory cytokines in relevant cell lines and human primary CD4+ T cells
A) THP-1 monocytic cells and phorbol ester-differentiated THP-1 macrophage cells were stimulated with 100 ng/mL LPS for 24 h while HT29 and Caco2 colonic epithelial cells were stimulated with 50 ng/mL and 5 ng/mL IL1ß, respectively, for 48 h to secrete pro-inflammatory cytokines. Cytokines were detected by bead-based flow cytometry. Activity of increasing concentration of rifamycin is expressed as Mean %inhibition + SEM relative to the 0.1% DMSO control (0% inhibition) B) Human CD4+ T cells were purified from peripheral blood mononuclear cells obtained from healthy volunteers. Cells were stimulated with 5 ng/mL phorbol ester and 500 ng/mL ionomycin for 24 h in the presence of 1 µM dexamethasone (Dex), 10 – 200 µM rifamycin, or DMSO then supernatants analyzed for cytokines using bead-based flow cytometry. The activity of the tested compounds is expressed as Mean %inhibition + SEM relative to the DMSO control (0% inhibition). °P < 0.05, °°P < 0.005, °°°P < 0.0005, °°°°P < 0.0005 vs. DMSO.
Several lines of evidence suggest that CD4+ T cells have a major contribution towards the pathogenesis of IBD38 and some evidence exists for their role in IBS39. Many subsets of CD4+ T cells have been identified as players in initiating and perpetuating chronic intestinal inflammation and several biologics that target CD4+ T cell differentiation into inflammatory subsets or their cytokines have shown efficacy in treating patients with IBD40. IBS patients have an increased frequency of colonic mast cells and activated CD4+ T cells, consistent with the hypothesis of low-grade immune activation which may also be involved in symptom generation in IBS39. Rifamycin was shown to inhibit IL2, IL17A, IFNγ, TNFα and IL6 from human primary CD4+ T cells that were induced to secrete these cytokines by treatment with phorbol ester (Figure 4B, modified from31). At the highest concentration tested, rifamycin inhibited all five cytokines by greater than 90% compared to the positive control steroidal anti-inflammatory agent, dexamethasone (Dex) at 1 µM, which inhibited up to 83% of secreted IL17A from these primary immune cells. This potent inhibition of several inflammatory cytokines from non-transformed primary human cells, in the same range as a steroidal drug, further support rifamycin as a therapeutic drug to treat mucosal inflammation in the context of IBD or IBS.
Concluding Remarks
The anti-inflammatory properties of rifamycin have been demonstrated for the first time in a study in which epithelial cells, exposed to sub-therapeutic concentrations of rifamycin showed a reduction in induced levels of inflammatory cytokines31. Importantly, this study also demonstrated that activated CD4+ T cells treated with rifamycin, secreted lower levels of cytokines that are critical to the pathogenesis of GI disorders. These results were followed up with a study to evaluate the activity of rifamycin on upstream regulators of these cytokines, namely PXR and NFκB. Indeed, in vitro studies in epithelial cell lines demonstrated that rifamycin also displays potent PXR- and NFκB-dependent anti-inflammatory activities25.
The reported in vitro results are clinically relevant based on several observations. First, the expression of PXR and its target genes are reduced in intestinal epithelial cells of patients with IBD21. Validation of PXR as a potential target for treatment of GI diseases in humans was demonstrated recently in various experimental GI disease models (reviewed in41), and reciprocal crosstalk between PXR and NFκB may be one of the mechanisms for the anti-inflammatory properties of PXR. Secondly, the use of TNF blocking drugs has firmly established the dogma that TNFα is a major pathological cytokine in IBD. TNFα-induced NFκB is particularly relevant not only in IBD, but also in IBS-D patients whose serum TNFα levels are significantly elevated compared to healthy subjects42, reviewed in43 and in patients with diverticular disease44. Our finding that rifamycin potently inhibits TNFα-induced NFκB supports its use in treating these patients. Moreover, since serum LPS is elevated in IBS-D patients45 the highly effective inhibition of LPS-induced NFκB by rifamycin further supports rifamycin as a therapeutic option for patients suffering from this GI disorder. Third, PXR is also involved in maintaining the integrity of the intestinal epithelial barrier by detoxification of luminal agents via induction of genes: CYP enzymes and transporters like PgP7, as well by maintenance of the physical barrier by expression of tight junction proteins in intestinal cells 6, 11. In some of the assays, rifamycin is better than rifampicin in stimulating PXR transcriptional activity, inducing PgP protein expression, and inducing NFκB activity. Thus, restoration of mucosal barrier integrity via anti-inflammatory and immunomodulatory activities of rifamycin, coupled with direct bactericidal properties, are expected to enhance the efficacy of this non-systemic antibiotic in the treatment of these GI inflammatory disorders. Lastly, the effective in vitro concentrations used in these studies are clinically achievable based on reported intestinal fluid volumes46 and the rifamycin dose used for traveler’s diarrhea30.
Restoration of intestinal homeostasis by rifamycin could potentially treat liver diseases as well through readjustment of the gut-liver axis. Compromised intestinal mucosal barrier exposes the liver to toxic factors derived from the intestines through the portal system, causing inflammation and hepatic injury. Indeed, experimental and clinical evidence implicate dysfunctions of this axis in the etiology of many liver diseases2. For example, the immune imbalances implicated in NAFLD pathophysiology are reported to stem from small intestinal bacterial overgrowth (SIBO), intestinal dysbiosis and increased permeability.
Despite experimental studies on the anti-inflammatory role of PXR in animal models, the exact clinical relevance in humans is not clear and needs further research because of the potential adverse effect on liver function after long-term exposure with PXR agonists12. In PXR-humanized mice, long-term stimulation of PXR by rifaximin, a non-absorbed antibiotic, resulted in hepatic steatosis due to up-regulation of intestinal genes involved in the uptake of fatty acids and triglycerides47. In addition, although PXR activation by rifampicin protected against colitis, it promoted growth factor-dependent colon cancer aggressiveness in an animal model48. In contrast, rifaximin inhibited the release of pro-angiogenic mediators in colon cancer cells through a PXR-dependent pathway49. These contradictory effects highlight the need for additional research to provide a better understanding of the long-term safety of PXR-targeted therapy for GI and liver diseases. Nevertheless, the positive attributes of rifamycin, which is chemically similar to rifampicin and rifaximin yet has functionally distinct properties from them, serve as encouraging points to pursue clinical use of this antibiotic for GI and liver malignancies.
Disclosure
Rifamycin was a gift from Cosmo Technologies Ltd, that sponsored and funded this study. The authors conducted the research summarized in this article and are currently employed by Cosmo Pharmaceuticals, the manufacturer of rifamycin.
References
- Camilleri M, Lyle BJ, Madsen KL, et al. Role for diet in normal gut barrier function: developing guidance within the framework of food-labeling regulations. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2019; 317: G17-G39.
- Wiest R, Albillos A, Trauner M, et al. Targeting the gut-liver axis in liver disease. Journal of Hepatology. 2017; 67: 1084-1103.
- Oladimeji PO, Chen T. PXR: More Than Just a Master Xenobiotic Receptor. Mol Pharmacol. 2018; 93: 119-127.
- Björkholm B, Bok CM, Lundin A, et al. Intestinal Microbiota Regulate Xenobiotic Metabolism in the Liver. PLOS ONE. 2009; 4: e6958.
- Terc J, Hansen A, Alston L, et al. Pregnane X receptor agonists enhance intestinal epithelial wound healing and repair of the intestinal barrier following the induction of experimental colitis. Eur J Pharm Sci. 2014; 55: 12-19.
- Venkatesh M, Mukherjee S, Wang H, et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity. 2014; 41: 296-310.
- Garg A, Zhao A, Erickson SL, et al. Pregnane X Receptor Activation Attenuates Inflammation-Associated Intestinal Epithelial Barrier Dysfunction by Inhibiting Cytokine-Induced Myosin Light-Chain Kinase Expression and c-Jun N-Terminal Kinase 1/2 Activation. J Pharmacol Exp Ther. 2016; 359: 91-101.
- Shin W, Kim HJ. Intestinal barrier dysfunction orchestrates the onset of inflammatory host–microbiome cross-talk in a human gut inflammation-on-a-chip. Proceedings of the National Academy of Sciences. 2018; 115: E10539-E10547.
- Rosenthal SB, Bush KT, Nigam SK. A Network of SLC and ABC Transporter and DME Genes Involved in Remote Sensing and Signaling in the Gut-Liver-Kidney Axis. Scientific Reports. 2019; 9: 11879.
- Sun M, Cui W, Woody SK, et al. Pregnane X receptor modulates the inflammatory response in primary cultures of hepatocytes. Drug metabolism and disposition: the biological fate of chemicals. 2015; 43: 335-343.
- Shimada Y, Kinoshita M, Harada K, et al. Commensal bacteria-dependent indole production enhances epithelial barrier function in the colon. PLoS One. 2013; 8: e80604.
- Shehu AI, Ma X. Pregnane X receptor in drug-induced liver injury: Friend or foe? Liver Research. 2018; 2: 173-179.
- Zhou C, Tabb MM, Nelson EL, et al. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest. 2006; 116: 2280-2289.
- Shah YM, Ma X, Morimura K, et al. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am J Physiol Gastrointest Liver Physiol. 2007; 292: G1114-1122.
- Mencarelli A, Migliorati M, Barbanti M, et al. Pregnane-X-receptor mediates the anti-inflammatory activities of rifaximin on detoxification pathways in intestinal epithelial cells. Biochem Pharmacol. 2010; 80: 1700-1707.
- Wallace K, Cowie DE, Konstantinou DK, et al. The PXR is a drug target for chronic inflammatory liver disease. The Journal of steroid biochemistry and molecular biology. 2010; 120: 137-148.
- Gu X, Ke S, Liu D, et al. Role of NF-kappaB in regulation of PXR-mediated gene expression: a mechanism for the suppression of cytochrome P-450 3A4 by proinflammatory agents. J Biol Chem. 2006; 281: 17882-17889.
- Pavek P. Pregnane X Receptor (PXR)-Mediated Gene Repression and Cross-Talk of PXR with Other Nuclear Receptors via Coactivator Interactions. Frontiers in Pharmacology. 2016; 7: 456.
- Luedde T, Schwabe RF. NF-κB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nature reviews. Gastroenterology & hepatology. 2011; 8: 108-118.
- Mackowiak B, Hodge J, Stern S, et al. The Roles of Xenobiotic Receptors: Beyond Chemical Disposition. Drug Metabolism and Disposition. 2018; 46:1361-1371.
- Langmann T, Moehle C, Mauerer R, et al. Loss of detoxification in inflammatory bowel disease: dysregulation of pregnane X receptor target genes. Gastroenterology. 2004; 127: 26-40.
- Han YM, Koh J, Kim JW, et al. NF-kappa B activation correlates with disease phenotype in Crohn’s disease. PLoS ONE. 2017; 12: e0182071.
- Ribeiro PS, Cortez-Pinto H, Sola S, et al. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol. 2004; 99: 1708-1717.
- He G, Karin M. NF-kappaB and STAT3 - key players in liver inflammation and cancer. Cell Res. 2011; 21: 159-168.
- Rosette C, Agan FJ, Rosette N, et al. Rifamycin SV exhibits strong anti-inflammatory in vitro activity through pregnane X receptor stimulation and NFkappaB inhibition. Drug Metab Pharmacokinet. 2019; 34: 172-180.
- Scarpignato C, Pelosini I. Rifaximin, a poorly absorbed antibiotic: pharmacology and clinical potential. Chemotherapy. 2005; 51 Suppl 1: 36-66.
- Farrell DJ, Putnam SD, Biedenbach DJ, et al. In vitro activity and single-step mutational analysis of rifamycin SV tested against enteropathogens associated with traveler's diarrhea and Clostridium difficile. Antimicrob Agents Chemother. 2011; 55: 992-996.
- Kogawa AC, Salgado HRN. Status of Rifaximin: A Review of Characteristics, Uses and Analytical Methods. Crit Rev Anal Chem. 2018; 48: 459-466.
- Hoy SM. Rifamycin SV MMX((R)): A Review in the Treatment of Traveller's Diarrhoea. Clin Drug Investig. 2019; 39: 691-697.
- Steffen R, Jiang ZD, Gracias Garcia ML, et al. Rifamycin SV-MMX(R) for treatment of travellers' diarrhea: equally effective as ciprofloxacin and not associated with the acquisition of multi-drug resistant bacteria. J Travel Med. 2018; 25.
- Rosette C, Buendia-Laysa F, Jr., Patkar S, et al. Anti-inflammatory and immunomodulatory activities of rifamycin SV. Int J Antimicrob Agents. 2013; 42: 182-186.
- Smutny T, Mani S, Pavek P. Post-translational and Post-transcriptional Modifications of Pregnane X Receptor (PXR) in Regulation of the Cytochrome P450 Superfamily. Current drug metabolism. 2013; 14: 1059-1069.
- Haslam IS, Jones K, Coleman T, et al. Induction of P-glycoprotein expression and function in human intestinal epithelial cells (T84). Biochem Pharmacol. 2008; 76: 850-861.
- Istrate MA, Nussler AK, Eichelbaum M, et al. Regulation of CYP3A4 by pregnane X receptor: The role of nuclear receptors competing for response element binding. Biochem Biophys Res Commun. 2010; 393: 688-693.
- Abualsunun WA, Piquette-Miller M. Involvement of Nuclear Factor kappaB, not Pregnane X Receptor, in Inflammation-Mediated Regulation of Hepatic Transporters. Drug Metab Dispos. 2017; 45: 1077-1083.
- Pan W, Yu C, Hsuchou H, et al. The role of cerebral vascular NFkappaB in LPS-induced inflammation: differential regulation of efflux transporter and transporting cytokine receptors. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2010; 25: 623-630.
- Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell. 2017; 168: 37-57.
- Li J, Ueno A, Iacucci M, et al. Crossover Subsets of CD4(+) T Lymphocytes in the Intestinal Lamina Propria of Patients with Crohn's Disease and Ulcerative Colitis. Dig Dis Sci. 2017; 62: 2357-2368.
- Ohman L, Isaksson S, Lindmark AC, et al. T-cell activation in patients with irritable bowel syndrome. Am J Gastroenterol. 2009; 104: 1205-1212.
- Imam T, Park S, Kaplan MH, et al. Effector T Helper Cell Subsets in Inflammatory Bowel Diseases. Frontiers in immunology. 2018; 9: 1212-1212.
- Pagliari D, Gambassi G, Piccirillo CA, et al. The Intricate Link among Gut "Immunological Niche," Microbiota, and Xenobiotics in Intestinal Pathology. Mediators Inflamm. 2017; 2017: 8390595.
- Rana SV, Sharma S, Sinha SK, et al. Pro-inflammatory and anti-inflammatory cytokine response in diarrhoea-predominant irritable bowel syndrome patients. Trop Gastroenterol. 2012; 33: 251-256.
- Bashashati M, Rezaei N, Shafieyoun A, et al. Cytokine imbalance in irritable bowel syndrome: a systematic review and meta-analysis. Neurogastroenterol Motil. 2014; 26: 1036-1048.
- Tursi A, Elisei W, Brandimarte G, et al. Musosal tumour necrosis factor alpha in diverticular disease of the colon is overexpressed with disease severity. Colorectal Dis. 2012; 14: e258-263.
- Dlugosz A, Nowak P, D'Amato M, et al. Increased serum levels of lipopolysaccharide and antiflagellin antibodies in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol Motil. 2015; 27: 1747-1754.
- Li J, Ueno A, Iacucci M, et al. Crossover Subsets of CD4(+) T Lymphocytes in the Intestinal Lamina Propria of Patients with Crohn's Disease and Ulcerative Colitis. Dig Dis Sci. 2017; 62: 2357-2368.
- Ohman L, Isaksson S, Lindmark AC, et al. T-cell activation in patients with irritable bowel syndrome. Am J Gastroenterol. 2009; 104: 1205-1212.
- Imam T, Park S, Kaplan MH, et al. Effector T Helper Cell Subsets in Inflammatory Bowel Diseases. Frontiers in immunology. 2018; 9: 1212-1212.
- Pagliari D, Gambassi G, Piccirillo CA, et al. The Intricate Link among Gut "Immunological Niche," Microbiota, and Xenobiotics in Intestinal Pathology. Mediators Inflamm. 2017; 2017: 8390595.
- Rana SV, Sharma S, Sinha SK, et al. Pro-inflammatory and anti-inflammatory cytokine response in diarrhoea-predominant irritable bowel syndrome patients. Trop Gastroenterol. 2012; 33: 251-256.
- Bashashati M, Rezaei N, Shafieyoun A, et al. Cytokine imbalance in irritable bowel syndrome: a systematic review and meta-analysis. Neurogastroenterol Motil. 2014; 26: 1036-1048.
- Tursi A, Elisei W, Brandimarte G, et al. Musosal tumour necrosis factor alpha in diverticular disease of the colon is overexpressed with disease severity. Colorectal Dis. 2012; 14: e258-263.
- Dlugosz A, Nowak P, D'Amato M, et al. Increased serum levels of lipopolysaccharide and antiflagellin antibodies in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol Motil. 2015; 27: 1747-1754.
- Schiller C, Frohlich CP, Giessmann T, et al. Intestinal fluid volumes and transit of dosage forms as assessed by magnetic resonance imaging. Alimentary Pharmacology & Therapeutics. 2005; 22: 971-979.
- Cheng J, Krausz KW, Tanaka N, et al. Chronic exposure to rifaximin causes hepatic steatosis in pregnane X receptor-humanized mice. Toxicological sciences : an official journal of the Society of Toxicology. 2012; 129: 456-468.
- Wang H, Venkatesh M, Li H, et al. Pregnane X receptor activation induces FGF19-dependent tumor aggressiveness in humans and mice. The Journal of clinical investigation. 2011; 121: 3220-3232.
- Esposito G, Nobile N, Gigli S, et al. Rifaximin Improves Clostridium difficile Toxin A-Induced Toxicity in Caco-2 Cells by the PXR-Dependent TLR4/MyD88/NF-kappaB Pathway. Front Pharmacol. 2016; 7: 120.