
With a Little Help from Good Friends – Boosters for the Prevention of Undesired Enzymatic Degradation of Anti-infective Drugs
Franz Bracher*
Ludwig-Maximilians University, Department of Pharmacy - Center for Drug Research, Butenandtstr., Munich, Germany
Abstract
The development of anti-infective drugs has been one of the most impressive progresses in drug therapy in the past century. However, some of the promising antibacterial and antiviral drugs lost activity after being used in therapy for some time. Typically, this is due to the development of resistance phenomena, among which the expression of drug-degrading enzymes is one major aspect. In other cases, enzymatic degradation of anti-infective drugs by mammalian enzymes in the liver (or kidney) can limit the efficacy of the drugs. In all of these cases, selection of a drug from a different class is a therapeutic opportunity. Alternatively, the original drug can be used further in combination with other compounds named boosters, pharmacokinetic enhancers or antibiotic adjuvants. These compounds are used in combination with the primary anti-infective agent, but not for their direct effects on the infection itself, but since they enhance or restore the activity of the drug. This mini-review gives an overview on the therapeutically most important classes of boosters/antibiotic enhancers, like β-lactamase inhibitors, inhibitors of CYP enzymes in HIV therapy and hepatitis C. Inhibitors of efflux pumps in pathogenic bacteria and fungi will be addressed shortly.
Introduction
The development of anti-infective drugs has been one of the most impressive progresses in drug therapy in the past century. However, we had to learn that some of the promising antibacterial, antifungal and antiviral drugs lost activity after being used in therapy for some time. Typically, this is due to the development of resistances, for which numerous molecular mechanisms have been detected, in other cases, enzymatic degradation of anti-infective drugs by mammalian enzymes can limit the efficacy of the drugs. In all of these cases, selection of an anti-infective drug from a different class is a therapeutic opportunity. Alternatively, the original drug can be used further in combination with other compounds named boosters, boosting agents, pharmacokinetic enhancers or antibiotic adjuvants1-2.
Boosters are substances which are used in combination with a primary therapeutic agent (drug), not for their direct effects on the pathogenic organisms or viruses themselves, but because they enhance or restore the activity of the anti-infective agent. So, in many cases they represent an indispensable escort for drugs, which otherwise would have to be discarded from therapeutic regimens.
A couple of reasons makes the application of boosters reasonable, some being associated with undesired enzymatic degradation by mammalian enzymes (most likely, hepatic drug-metabolizing enzymes such as CYP (cytochrome P450) 3A4, but also other drug-specific metabolizing enzymes, e.g. in the kidney), others with drug-degrading enzymes in (mutated) pathogenic microorganisms (e.g., bacterial β-lactamases). It should be noted that a couple of other reasons may be responsible for development of resistances of pathogenic microorganisms, like mutation of target proteins and expression of efflux pumps which eliminate the anti-infective drugs from the microbial cells, but these phenomena are poorly addressable by boosters and will be treated here only shortly1.
A comprehensive review on boosters (pharmacokinetic enhancers), also including applications in other fields like treatment of tumors and Morbus Parkinson, with a stronger focus on drug structures and molecular modes of actions, has been published recently3. This mini review will focus on the presently launched boosters utilized for co-administation with anti-infective drugs. Compounds in late stages of clinical development are mentioned in special cases.
Boosters for Antibiotic Chemotherapy
Boosters targeting bacterial enzymes: β-lactamase inhibitors
The β-lactams (penicillins, cephalosporins, carbapenems, monobactams, and others) are one of the most important classes of antibiotics, but resistance to β-lactam antibiotics emerged to a severe problem in anti-infective therapy over the decades. Due to the broad and (in part) poorly controlled application of antibiotics, this problem is increasing dramatically4. Unfortunately, due to the poor rate of development of novel antibiotics with alternative molecular modes of action, it is very hard to fill all of the emerging therapeutic gaps. Consequently, preserving the therapeutic utility of “old” antibiotics with “resistance breakers” (also called potentiators) is a most attractive strategy5. These agents are applied in combination with the vulnerable antibiotic, and their dominating function is to protect the antibiotics from enzymatic degradation by bacterial enzymes. Typically, these agents do not display own antibiotic activity, and consequently, they cannot be administered without the antibiotic partner1.
The most prominent example is inactivation of β-lactam antibiotics by bacterial β-lactamases. These enzymes are able to cleave the crucial four-membered β-lactam ring of the antibiotics by formal amide hydrolysis to give inactive, ring-opened metabolites1,6. The β-lactam ring is the active motif of these antibiotics, binding to the bacterial target proteins (penicillin-binding proteins, transpeptidases) required for the biosynthesis of the bacterial cell wall component murein. In this course, the β-lactam antibiotics bind to a serine hydroxyl group in the active site of the bacterial enzymes, leading to irreversible inactivation (Figure 1, left). Bacterial serine-β-lactamases also contain a serine hydroxyl group in the active site, cleave the β-lactam ring of the antibiotics, but then are released in their active form from the inactive fragment of the antibiotic, and so can catalyze destruction of numerous antibiotics molecules in the following, leading to a dramatic loss of active antibiotic (Figure 1, center). The activity of the labile β-lactam antibiotic can be restored by co-application of a β-lactamase inhibitor, like clavulanic acid, sulbactam and tazobactam (Figure 1, right). These boosters, which themselves contain a β-lactam structure, inactivate the bacterial serine β-lactamases and thus inhibit enzymatic degradation of the β-lactam antibiotics. They are called mechanism-based covalent inhibitors, since they react irreversibly with the active site serine hydroxyl group of the β-lactamases under formation of stable acyl–enzyme complexes leading to inactivation of the enzyme, as exemplified for clavulanic acid here.
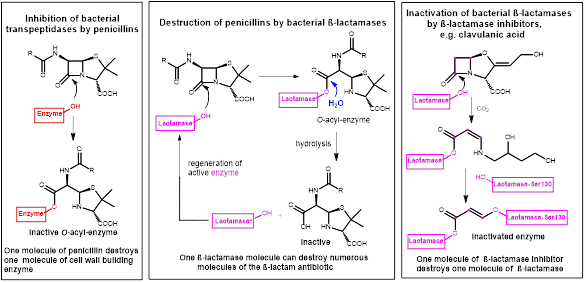
Figure 1. Left: β-lactam antibiotics like penicillins form a stable O-acyl-enzyme complex by reaction with a serine residue in the active site of the bacterial target enzyme. Center: Hydrolysis and inactivation of β-lactam antibiotics by bacterial β-lactamases; active β-lactamase is regenerated here. Right: β-lactamase inhibitors inactivate the β-lactamase enzyme by forming covalent bonds in the active site.
So, β-lactamase inhibitors are the most widely clinically used adjuvants to overcome resistance caused by β-lactamases. Typical launched combinations are clavulanic acid/amoxicillin for oral application, whereas tazobactam/piperacillin and sulbactam/ampicillin combinations are highly established in parenteral therapy.
But it has to be noted that β-lactamases represent a large and heterogeneous class of hydrolyzing enzymes with over 500 reported representatives, so co-administration of one of the launched by β-lactamase inhibitors does by far not guarantee a striking antibiotic effect1. For example, these first generation β- inhibitors lack activity against so-called class C and D-type β-lactamases.
Major research has been performed to overcome these shortcomings, and in the past few years a novel chemotype of β-lactamase inhibitors has been developed, which in part overcomes these limitations, with avibactam as the pioneering substance, launched in combination with the cephalosporin ceftazidime6-7. This inhibitor contains a bridged γ-lactam ring (meaning: a five-membered lactam instead of the four-membered one found in the antibiotics and the first generation β-lactamase inhibitors shown in Figure 2). Avibactam also binds covalently to the serine residue in the active site of the β-lactamases, but in contrast to the binding mode of first-generation inhibitors, this binding is reversible. Nevertheless, this leads to an effective inhibition of β-lactamase activity. A closely related inhibitor, relebactam, has been approved in 2019 in the USA in combination with imipenem and cilastatin (see next chapter).
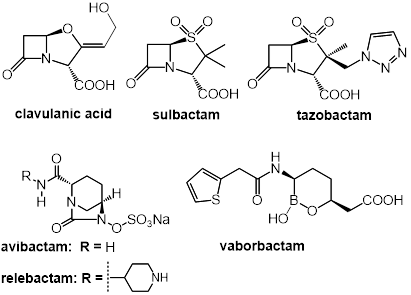
Figure 2. Structures of β-lactamase inhibitors. Top: the β-lactam-type inhibitors clavulanic acid, sulbactam, and tazobactam. Bottom: the bridged γ-lactams avibactam and relebactam, and the boronic acid-derived inhibitor vaborbactam. While the β-lactam- and γ-lactam-type inhibitors destroy β-lactamases via formation of O-acyl enzyme intermediates (see Figure 1, right), the boronic acid derivative vaborbactam forms boronate adduct with the active site serine hydroxyl group.
As described above, both the β-lactam and γ-lactam-type serine β-lactamase inhibitors lead to enzyme inhibition by chemical reactions with serine hydroxyl groups in the active site of the β-lactamases. A mechanistically novel type of β-lactamase inhibitors are the so-called non-acylating inhibitors, which do not contain a reactive lactam ring for binding to the active site of the enzymes. In contrast, these new β-lactamase inhibitors, e.g. vaborbactam, contain a boronic acid-derived functionality which undergoes a reversible interaction with the active site serine hydroxyl group by forming a tetrahedral, negatively charged boronate adduct. High affinity for β-lactamases is achieved in this molecule by incorporation of side chains with close structural similarity to the penicillin-type substrates of the enzymes. Vaborbactam, launched in 2017, effectively blocks clinically relevant class A and C β-lactamases, including carbapenemases, and and is used in combination with the carbapenem antibiotic meropenem1,8. A couple of related boronate-based β-lactamase inhibitors are presently in clinical trials.
A booster targeting a mammalian host enzyme: cilastatin
The carbapenem-subtype of β-lactam antibiotics are very important broad-spectrum antibiotics. Especially imipenem (Figure 3) has high clinical relevance for treatment of infections with Pseudomonas aeruginosa and Enterococci. Further, imipenem has for a long time been rather resistant to critical β-lactamases which meanwhile shrink the therapeutic use of other β-lactam antibiotics (penicillins, cephalosporins). However, imipenem is subject to significant enzymatic degradation by the mammalian renal enzyme dehydropeptidase-I, and for this reason cannot be used as a monotherapy. Dehydropeptidase-I degrades imipenem in a mechanism similar to the action of bacterial β-lactamasaes, by hydrolyzing the β-lactam ring to an inactive metabolite (compare Figure 1 showing the analogous degradation of penicillins). Antibiotic therapy with imipenem could only be made available after development of a potent inhibitor of human dehydropeptidase-I, and in a systematic search and optimization process cilastatin was developed as a booster for imipenem (Figure 3)9-10. Like the β-lactamase inhibitors described above, cilastatin does not have own antibacterial activity.
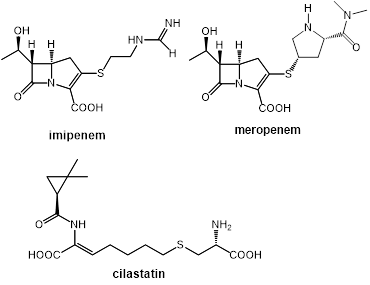
Figure 3. Structures of the carbapenem-type antibiotics imipenem and meropenem. Imipenem needs boosting by the dehydropeptidase-I inhibitor cilastatin, whereas meropenem is not a substrate of the renal enzyme, and can be applied without this booster. Meanwhile, both antibiotics need boosting with β-lactamase inhibitors.
Surprisingly, the closely related carbapenem antibiotic meropenem (launched in fixed combination with the novel boronic acid-derived β-lactamase inhibitor vaborbactam) is not a substrate of renal dehydropeptidase-I11. As meanwhile also imipenem-degrading β-lactamases (carbapenemases) came up, a combination of imipenem with both cilastatin and the novel β-lactamase inhibitor relebactam1 (see above) has been approved in the USA in 2019.
Boosters in HIV therapy: Inhibition of CYP3A4 by ritonavir and cobicistat for boosting antiretroviral drugs
A broad spectrum of antiretroviral drugs with multiple molecular modes of action have been developed over the decades for controlling HIV infections. Efficient and durable suppression of the virus load is commonly achieved with novel therapy regimens implying permanent therapy with three or more antiretroviral drugs from different classes like entry inhibitors, reverse transcriptase inhibitors, integrase inhibitors and, most prominently, protease inhibitors. The enzyme HIV protease catalyzes controlled hydrolysis of viral peptidic precursors into the required enzymes and structural proteins. Initial poor absorption after oral administration of the early peptidic protease inhibitors was overcome by sophisticated design of lipophilic peptidomimetic analogues like ritonavir, lopinavir, atazanavir, and darunavir. Nevertheless, poor bioavailability of these protease inhibitors remained a major challenge for a different reason: These drugs are effectively inactivated by CYP3A4-catalyzed oxidation in a first pass metabolism in the gut wall during absorption, and in the liver. Consequently, boosting the effect of protease inhibitors is effectively achieved with CYP3A4 inhibitors12-13.
Notably, ritonavir (Figure 4), a peptidomimetic substance originally developed and launched as a HIV protease inhibitor, was found to be a potent CYP3A4 inhibitor as well, and meanwhile it is, in lower doses, a standard component of multidrug regimens in this field, mainly since it acts as a booster for CYP3A4-labile co-administered drugs, preventing their metabolism and allowing to use well-tolerated lower doses and more patient-friendly regimens of these drugs14. Fortunately, ritonavir further blocks P-glycoprotein (Pgp) efflux transporters which pump numerous antiretroviral drugs out of the gut wall back into the intestinal lumen, and so reduce oral bioavailabiltity of the drugs further12-13. This boosting effect is not limited to protease inhibitors. The HIV integrase inhibitor elvitegravir (Figure 4) is also protected from oxidative degradation by ritonavir as well as its recently developed analogue cobicistat. Notably, other launched integrase inhibitors (raltegravir, dolutegravir), are not degraded significantly by CYP3A4, so for these drugs boosting is not required15.
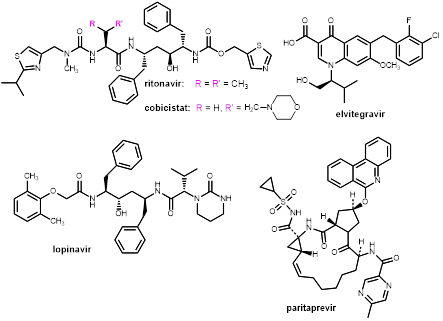
Figure 4. Structures of the boosters (CYP inhibitors) ritonavir and cobicistat, and selected antiviral drugs boosted by these compounds through CYP inhibition: the HIV integrase inhibitor elvitegravir, the HIV protease inhibitors lopinavir, and the HCV NS3/4A inhibitor paritaprevir.
Cobicistat (Figure 4), launched in 2012 as Tybost® for combination with HIV protease inhibitors and the integrase inhibitor elvitegravir, does, in contrast to ritonavir, not show own inhibition of HIV protease, it is a pure booster blocking CYP3A4 and Pgp efflux pumps13-14. Cobicistat has been claimed to have fewer side effects and a better CYP inhibition profile than ritonavir14,16, but recent investigations suggest that there are no striking differences between both compounds in clinical value17.
Ritonavir as a booster in hepatitis C therapy
Recently, ritonavir found application in antiviral therapy beyond HIV. Hepatitis C is a severe, life-threatening viral infection, and only in the past few years, novel so-called direct-acting antivirals (DAAs) have been developed which revolutionized hepatitis C therapy. Among these medicines is the fixed combination of the NS3/4A inhibitor paritaprevir, the NS5A inhibitor ombitasvir and ritonavir (Viekirax®, TechnivieTM). For treatment of HCV1 genotype-1 patients this is further combined with the NS5B polymerase inhibitor dasabuvir18. One of these components, paritaprevir (Figure 4), is a potent competitive inhibitor of the HCV serine protease NS3 and its co-factor NS4A. Paritaprevir is a substrate of CYP3A4, and thus co-administration of the booster ritonavir is mandatory for reaching sufficient plasma levels of paritaprevir. In contrast to HIV therapy (see above), ritonavir does not have inhibitory activity on HCV proteases, and it is a pure booster for paritaprevir here. The other active components in this combination therapy are not substrates of CYP3A4, and consequently do not need boosting16.
Addendum: Boosting anti-infective drugs by interaction with human and microbial transport processes
The boosters described above act as enzyme inhibitors, thus block/reduce undesired metabolism and inactivation of the anti-infective drug, leading to an increase in available concentration of the active agent. In principle, the available concentration of the drug (either in the microbial cell to be killed or in the relevant human organ/tissue) can also be increased by blocking mechanisms which remove the drug from the place where it is needed. In the 1940s of the past century, probenecid (Figure 5), a simple sulfonamide-derived blocker of the organic anion transporter, was used in combination with penicillins, since it significantly reduces the renal excretion of penicillins, and helped to save the valuable antibiotics19. Later similar effects on the renal excretion were detected for a number (but not all!) fluoroquinolone antibiotics, like ciprofloxacin and levofloxacin20. Presently, probenecid is utilized in combination with cidofovir (Figure 5), an antiviral nucleotide analogue used for treatment of cytomegalovirus retinitis in AIDS patients. Cidofovir is renally eliminated as the unchanged drug, whereby an "organic anion transporter" located on the basolateral side of the tubular cells actively pumps the drug into the tubular cells. The subsequent secretion into the tubular lumen takes place at a significantly lower rate, leading to an accumulation of cidofovir in the proximal tubular cells. This is, most likely, the reason for the well-documented nephrotoxicity of cidofovir21. Blocking this anion transporter with probenecid helps to avoid this accumulation in the tubular cells and reduces this toxic side-effect. Since also the overall renal clearance of cidofovir is blocked this way, this co-administration also leads to increased serum concentrations of the drug22.
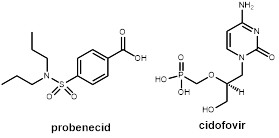
Figure 5. Probenecid, a blocker of the organic anion transporter, reduces renal elimination of certain drugs. It is combined with the antiviral drug cidofovir for reducing nephrotoxicity of this drug.
Significant research has also been performed for developing inhibitors of bacterial efflux pumps (multidrug resistance (MDR) efflux pumps). These pumps are an important reason for intrinsic and acquired resistance of bacteria, most prominently found in Gram-negative bacteria, since they eliminate anti-infective drugs from the microbial cells most effectively and fast, leading to insufficient activity of the antibiotics23. Despite numerous candidate compounds published, and encouraging in vitro data, none of these compounds has been launched yet, either due to missing in vivo activity or intolerable side effects. The same holds for fungal efflux pumps, e.g. of Candida species, also here no booster reached the market yet24.
Conclusion
The boosters (antibiotic enhancers) described here are valuable compounds which, when co-administered with an anti-infective drug can significantly enhance the therapeutic effect. These well-designed substances inhibit distinct enzymes, either in the resistant microorganisms or in human body, and thus inhibit (or at least strongly reduce) undesired metabolism and inactivation of the drug, leading to an increase in available concentration of the active agent and prolongation of its half-life13. Certain anti-infective drugs can only be utilized for therapy by applying this therapeutic concept, especially in case of pathogenic microorganisms which exhibit resistance to drugs on the basis of expression of drug degrading enzymes, like β-lactamases. Further, this concept typically allows lower dosage of the active component and/or reduced dosing frequency, both of them means for improvement of the medication adherence of patients.
However, development of perfect boosters is a complex task. As these boosters act as a kind of escort for the active drug, it is mandatory that they have comparable pharmacokinetics – only then they will be present in the same compartment of the human body (place of infection) over the entire time anti-infective activity has to be maintained. Further, the boosters should, if ever possible, not exert own, undesired pharmacological effects, and should not interfere with other drugs the patient is using. Especially for boosters which act by inhibiting CYP enzymes, this goal is hard to reach, and careful control has to be taken in order to detect undesired interactions with the metabolism of the other drugs in multimorbid patients.
References
- González-Bello C. Antibiotic adjuvants – A strategy to unlock bacterial resistance to antibiotics. Bioorg Med Chem Lett. 2017; 27: 4221-4228.
- Gill EE, Franco OL, Hancock RE. Antibiotic adjuvants: diverse strategies for controlling drug-resistant pathogens. Chem Biol Drug Design. 2015; 85: 56-78.
- Krauß J, Bracher F. Pharmacokinetic Enhancers (Boosters)—Escort for Drugs Against Degrading Enzymes and Beyond. Sci Pharm. 2018; 86: 43.
- Cassini A, Hogberg LD, Plachouras D, et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: a population-level modelling analysis. Lancet Infect Dis. 2019; 19: 56-66.
- Chakradhar S. What's old is new: Reconfiguring known antibiotics to fight drug resistance. Nature Med. 2016; 22: 1197-1199.
- Wang DY, Abboud MI, Markoulides MS, et al. The road to avibactam: the first clinically useful non-β-lactam working somewhat like a β-lactam. Fut Med Chem. 2016; 8: 1063-1084.
- Shirley M. Ceftazidime-Avibactam: A Review in the Treatment of Serious Gram-Negative Bacterial Infections. Drugs. 2018; 78: 675-692.
- Hecker SJ, Reddy KR, Totrov M, et al. Discovery of a Cyclic Boronic Acid β-Lactamase Inhibitor (RPX7009) with Utility vs Class A Serine Carbapenemases. J Med Chem. 2015; 58: 3682-3692.
- Keynan S, Hooper NM, Felici A, et al. The renal membrane dipeptidase (dehydropeptidase I) inhibitor, cilastatin, inhibits the bacterial metallo-beta-lactamase enzyme CphA. Antimicrob Agents Chemother. 1995; 39: 1629-1631.
- Humanes B, Jado JC, Camaño S, et al. Protective Effects of Cilastatin against Vancomycin-Induced Nephrotoxicity. BioMedRes Intern. 2015; Article ID 704382.
- Fukasawa M, Sumita Y, Harabe ET, et al. Stability of meropenem and effect of 1 beta-methyl substitution on its stability in the presence of renal dehydropeptidase I. Antimicrob Agents Chemother. 1992; 36: 1577-1579.
- Larson KB, Wang K, Delille C, et al. Pharmacokinetic enhancers in HIV therapeutics. Clin Pharmacokinet. 2014; 53: 865-872.
- Renjifo B, van Wyk J, Salem AH, et al. Pharmacokinetic enhancement in HIV antiretroviral therapy: a comparison of ritonavir and cobicistat. AIDS Reviews. 2015; 17: 37-46.
- Tseng A, Hughes CA, Wu J, et al. Cobicistat Versus Ritonavir: Similar Pharmacokinetic Enhancers But Some Important Differences. Ann Pharmacother. 2017; 51: 1008-1022.
- Atta MG, De Seigneux S, Lucas GM. Clinical Pharmacology in HIV Therapy. Clin J Am Soc. Nephrol. 2018; 10.2215/cjn.02240218.
- Hussaini T. Paritaprevir/ritonavir-ombitasvir and dasabuvir, the 3D regimen for the treatment of chronic hepatitis C virus infection: a concise review. Hepat Med. 2016; 8: 61-68.
- Hossain MA, Tran T, Chen T, et al. Inhibition of human cytochromes P450 in vitro by ritonavir and cobicistat. J Pharm Pharmacol. 2017; 69: 1786-1793.
- Ahmed A, Felmlee DJ. Mechanisms of Hepatitis C Viral Resistance to Direct Acting Antivirals. Viruses. 2015; 7: 6716-6729.
- Robbins N, Koch SE, Tranter M, et al. The history and future of probenecid. Cardiovasc Toxicol. 2012; 12: 1-9.
- Landersdorfer CB, Kirkpatrick CM, Kinzig M, et al. Competitive inhibition of renal tubular secretion of ciprofloxacin and metabolite by probenecid. Brit J Clin Pharmacol. 2010; 69: 167-178.
- Cundy KC, Petty BG, Flaherty J, et al. Clinical pharmacokinetics of cidofovir in human immunodeficiency virus-infected patients. Antimicrob Agents Chemother. 1995; 39: 1247-1252.
- Uwai Y, Ida H, Tsuji Y, et al. Renal Transport of Adefovir, Cidofovir, and Tenofovir by SLC22A Family Members (hOAT1, hOAT3, and hOCT2). Pharm Res. 2007; 24: 811-815.
- Spengler G, Kincses A, Gajdacs M, et al. New Roads Leading to Old Destinations: Efflux Pumps as Targets to Reverse Multidrug Resistance in Bacteria. Molecules. 2017; 22.
- Holmes AR, Cardno TS, Strouse JJ, et al. Targeting efflux pumps to overcome antifungal drug resistance. Fut Med Chem. 2016; 8: 1485-1501.